Introduction to Whole Transcriptome Sequencing

Whole Transcriptome Sequencing allows characterization of all types of RNA transcripts (coding and non-coding RNAs) of a particular organism, irrespective of whether or not they are polyadenylated. Whole transcriptome sequencing is advocated for both differential expression analysis as well as discovery work.
Novogene’s Whole Transcriptome Sequencing service equips the researchers with cutting-edge NGS solutions that provide in-depth bioinformatic analyses of all RNA transcripts including mRNA and non-coding RNA (lncRNA, sRNA, and circRNA). By using this paired-end sequencing approach, we can precisely quantify gene and transcript levels, identify splice variants and novel characteristics of the transcriptome. This competitive approach investigates and explores potential transcriptional and regulatory network mechanisms while providing key insights into the interaction functionalities from a comprehensive transcriptomic perspective.
Applications of Whole Transcriptome Sequencing
Whole Transcriptome Sequencing finds its applications in:
- Profiling mRNA and ncRNA in a single run
- Exploring miRNA sponge and target regulatory elements
- Investigating regulatory networks among lncRNA/circRNA-miRNA-gene pairs
Benefits of Whole Transcriptome Sequencing
- Whole Transcriptome Sequencing provides a more comprehensive analysis of transcriptional regulation network, compared to mRNA-seq, lncRNA-seq, sRNA-seq, and circRNA-seq, respectively.
- Whole transcriptome sequencing helps researchers in identifying biomarkers across a wide transcript range.
- WTS allows the capture of both known and new features.
- It enables the whole transcriptome profiling across a broad dynamic scale.
Latest Blogs
eBooks
Do Transcriptome Analysis With Just a Click
Transcriptome sequencing, also referred to as RNA sequencing(RNA-seq) or massively parallel RNA-seq, is a powerful tool that offers deep insights into gene expression and how changes in gene expression drive biological processes and impact health. However, RNA-seq experiments generate large amounts of data that must be carefully managed and analyzed. RNA-seq data analysis is a complex pipeline requiring different types of data processing, quality control (QC), and statistical analysis at each stage. Naturally, those necessary analyses require specialized software tools. Here, we briefly discuss several of the analytical steps involved, and how you can use the tools of Novogene’s NovoMagic platform to accelerate your RNA-seq data analysis.
WTS Specifications: RNA Sample Requirements
| Library Type | Sample Type | Amount | RNA Integrity Number (Agilent 2100) |
Purity (NanoDrop) |
| lncRNA Library & small RNA Library | Total RNA | ≥ 3 μg | ≥ 7.5, with flat baseline (animal) ≥ 7.0, with flat baseline (plant, fungi) |
0D260/280≥2.0; 0D260/230≥2.0; no degradation, no contamination |
WTS Specifications: Sequencing and Analysis
| Sequencing Platform | Illumina NovaSeq 6000 Sequencing System |
| Read length | Paired-end 150bp for lncRNA/circRNA library
Single-end 50bp for small RNA library |
| Recommended Data Amount | ≥ 40 million read pairs per sample (lncRNA library); 10-20 million reads per sample (small RNA library); |
| Content of Data Analysis |
|
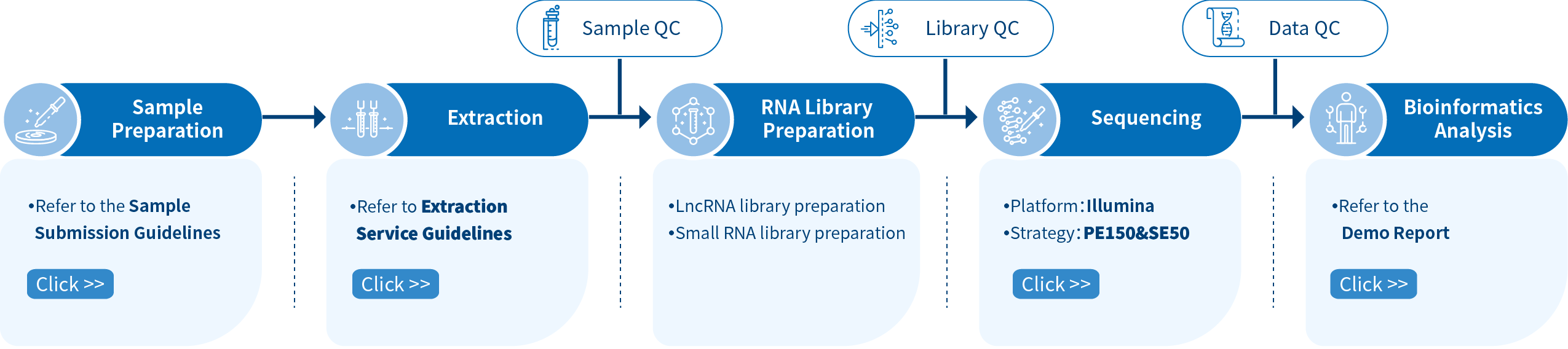
Novogene Workflow of Whole Transcriptome Sequencing Service
The first step of the project workflow includes the sample quality control (Sample QC) to ensure that your samples meet the criteria of the RNA-Seq technique. Then, the appropriate library is prepared according to your target organism and subsequently tested for its quality(Library QC). Next, a paired-end 150 bp sequencing strategy is used to sequence the lncRNA and circRNA library and single-end 50bp is used to sequence the small RNA library. The resulting data go through quality data control (Data QC) to guarantee the quality of the resulting data. Finally, bioinformatic analyses are performed and publication-ready results are provided. The following flowsheet describes the step-by-step protocol.
Featured Publications
Case Studies



Correlation analysis of circRNA and miRNA and mRNA
The circRNA-miRNA-gene triplets in which circRNA, miRNA, and genes are all differentially expressed were selected, and their numbers were counted in each comparison.
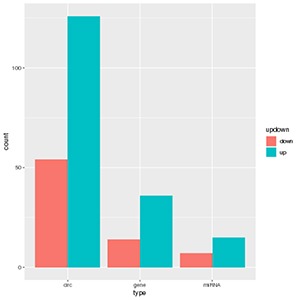
Figure 1 circRNA-miRNA-gene counting and networks
Note:
The histogram graph shows the number of differentially expressed genes, circRNA, miRNA in lncRNA-miRNA-gene triplets in each comparison
Interaction network of circRNA-miRNA-gene
circRNA harbors miRNA binding sites and acts as a molecular sponge for a miRNA to competitively keep miRNA from suppressing its downstream target genes of the corresponding miRNA family. Based on the competitive endogenous RNA (ceRNA) hypothesis, we filter out circRNA and genes targeted by the same miRNA and construct ceRNA regulation networks to reveal the expression regulation mechanisms of circRNA at the whole transcriptome level.
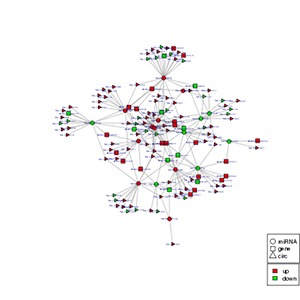
Note:
In the figure, different shapes represent different RNA types, and different colors represent the up-and down-regulation of the RNA genes. The size of a node is proportional to its degree. If more lines are connected to a node, its degree will be greater, and correspondingly the size will also be larger. These nodes are more likely to be in a core position in the network.
*Please contact us to get the full demo report.