Introduction to Microbial De novo Sequencing
De novo sequencing can sequence the genome of a species without any reference genome information, splice and assemble it by bioinformatics analysis methods, and obtain the genome sequence map of the species, to promote the follow-up research of the species. It offers reference genome assembly for rarely studied species. Using de novo sequencing to obtain the genomic information of microbes provides a fresh start for exploring the genetic structure and functions, studying the evolutionary origin of microbial populations, as well as developing potential applications of these abundant microbes in medicine, disease, agriculture, and the environment.
Novogene offers de novo sequencing services using both PacBio and Illumina platforms. We provide multifaceted sequencing services including genome survey, draft map, complete map, and fine map tailored to different research needs. For each project, our scientists will design the best sequencing strategy utilizing an optimal combination of short reads and long-range sequencing information to achieve the most comprehensive de novo assembly results for your genome of interest.
Applications of Microbial De novo Sequencing
For individual research:
- Virulence research
- Drug resistance mechanism
- Molecular markers
- Vaccine development
For population research:
- Evolution relationship
- Population size
- Epidemiology
- Microbial evolution
Benefits of Microbial De novo Sequencing
- Highly experienced: We have completed numerous microbial De novo sequencing projects with 20 publications in top-tier journals.
- Largest sequencing capacity: We have the largest Illumina and PacBio sequencing capacity in the world, allowing us to provide high-quality data, fast turnaround, and affordable prices.
- n-depth data mining: We have complete solutions for in-depth analysis of individual variations and interpretation of population evolutionary history to solve your biological problems that interest you.
- Accuracy: Creates accurate reference sequences, even for complicated or polyploid genomes.
Microbial De novo Sequencing Specifications:
DNA Sample Requirements
| Platform Type | Sample Type | Amount (Qubit®) | Purity |
| Illumina
NovaSeq 6000 |
Genomic DNA | ≥ 200 ng | A260/280=1.8-2.0 no degradation, no contamination |
| PacBio Revio DNA HiFi library | HMW Genomic DNA (Bacteria and Fungus) |
≥ 5 μg (Concentration ≥ 70 ng/μL) |
A260/280=1.75-2.0; A260/230=1.3-2.6; *NC/QC=1.0-2.2 Fragments should be ≥ 20 kb |
| PacBio PCR product library |
PCR product (Fungus) | ≥ 2 μg | OD260/280=1.75~2.0; OD260/230=1.4~2.6; *NC/QC=0.95~3.00; Single band (PacBio library fragments distributed above 1k) |
| Nanopore PromethION | HMW Genomic DNA (Bacteria and Fungus) |
≥ 6 μg (Concentration ≥ 60 ng/μL) |
A260/280=1.7-2.2; A260/230=1.3-2.6; *NC/QC=0.95-3.00 Fragments should be ≥ 20 kb |
| Nanopore PCR product library |
PCR product (Bacteria and Fungus) | ≥ 2 μg | OD260/280=1.75~2.0; OD260/230=1.4~2.6; *NC/QC=0.95~3.00; Single band |
* NC/QC: NanoDrop concentration/Qubit concentration
Microbial De novo Sequencing Specifications:
Sequencing & Analysis
| Platform Type | Illumina NovaSeq | PacBio Sequel II/IIe System | |
| Read Length | Paired-end 150 bp | N50>15 kb, long read lengths up to 25 kb(CCS) | |
| Recommended Sequencing Depth | ≥ 50x for bacterial and fungal genomes | ≥ 100X for bacterial genomes ≥ 50X for fungal genomes | |
| Data Analysis |
|
Repeat annotation Coding gene annotation ncRNA annotation GO annotation, KEGG annotation, COG annotation NR annotation, TCDB annotation Pfam annotation Swiss-Prot annotation
|
Repeat annotation Coding gene annotation ncRNA annotation GO annotation, KEGG annotation, NR annotation, KOG annotation Pfam annotation Swiss-Prot annotation. |
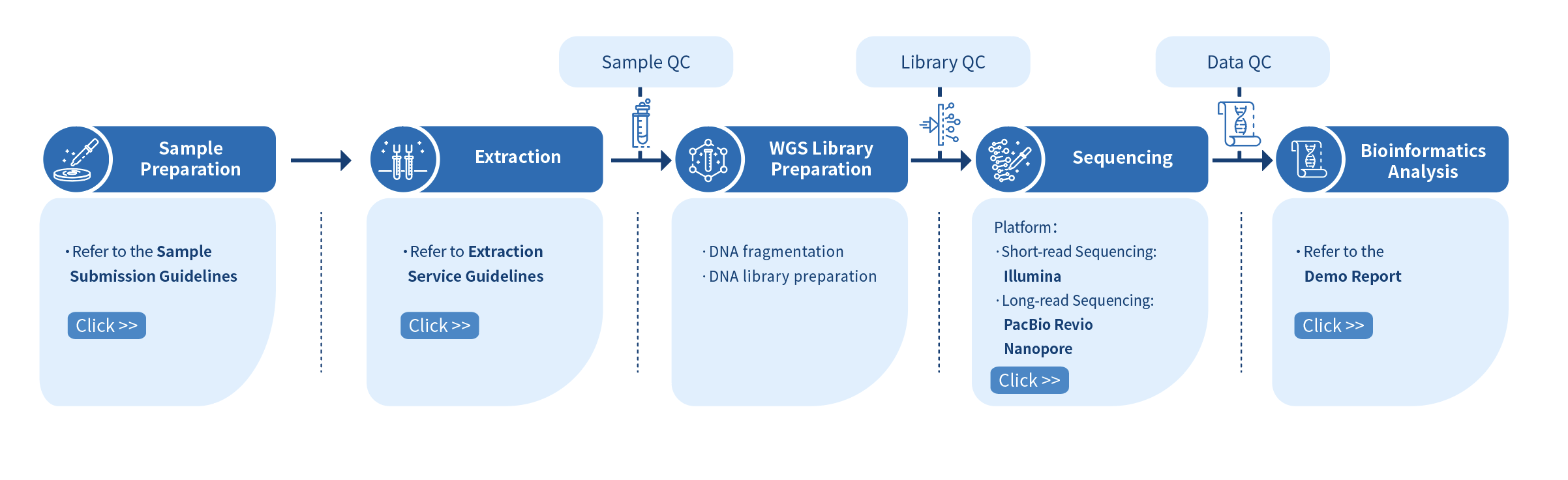
Novogene Workflow of Microbial De novo Sequencing
The first step of the project workflow involves the sample quality control (Sample QC) to ensure that your samples meet the criteria of the Microbial De novo Sequencing. Then, the appropriate library is prepared according to your target organism and application and subsequently tested for its quality (Library QC). Next, the sequenced sample and the resulting data are also checked for their quality (Data QC). Finally, bioinformatic analyses are performed and publication-ready results are provided. The following flowsheet describes the step-by-step protocol our Microbial De novo Sequencing follows.
Featured Publications of Microbial De novo Sequencing
-
Whole genome sequence of Diaporthe capsici, a new pathogen of walnut blight
Genomics Date: 23 February 2021IF: 6.205DOI: https://doi.org/10.1016/j.ygeno.2020.04.018
-
ISME Date: 14 September 2020IF: 9.493DOI: https://10.1038/s41396-020-0684-5
-
Scientific Reports Date: 31 August 2017IF: 5.228DOI: https://10.1038/s41598-017-10376-0
Long Read Sequencing (aiming at Bacteria Complete Map and Fungus Fine Map)
Genome Assembly
Polymerase read statistics
Polymerase reads are mostly used for quality control of the instrument run. Polymerase read metrics primarily reflect movie length and other run parameters rather than insert size distribution. Polymerase reads are trimmed to include only the high-quality region. Note: Sample quality is a major factor to be considered in polymerase read metrics.
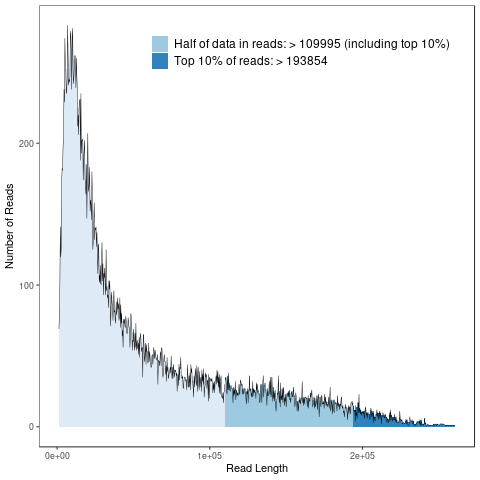
Polymerase read length distribution
Note:
The horizontal axis shows polymerase read length distribution, the vertical axis shows the number of reads corresponding to length distribution.
Subreads statistics
Each polymerase read is partitioned to form one or more subreads, which contain a sequence from a single pass of a polymerase on a single strand of an insert within a SMRTbell template and no adapter sequences. The subreads contain the full set of quality values and kinetic measurements. Subreads are useful for applications such as de novo assembly, resequencing, and base modification analysis.
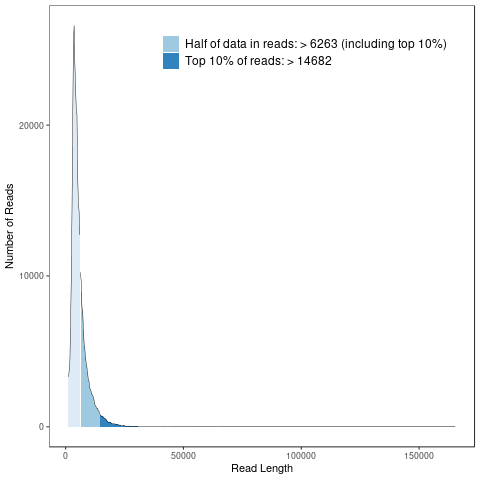
Subreads read length distribution
Note:
The horizontal axis shows subreads length distribution, the vertical axis shows the number of reads corresponding to length distribution.
Assembled genome statistics
19G PacBio raw reads were generated and used for assembly. The assembled genome sequence statistics information is shown in table
Assembled Genome Statistics
| Title | Total_length | Total_number | Num>=2000 | Average_length | Max_length | N50_length | N50_number | N90_length | N90_length |
| Contig | 3,024,820 | 1 | 1 | 3,024,820 | 3,024,820 | 3,024,820 | 1 | 3,024,820 | 1 |
| Scaffold | 3,024,820 | 1 | 1 | 3,024,820 | 3,024,820 | 3,024,820 | 1 | 3,024,820 | 1 |
Gene function annotation
GO Annotation
GO stands for Gene Ontology. The Gene Ontology (GO) project aims to provide reliable descriptions of gene products within several databases. GO vocabularies (ontologies) explain gene products concerning their associated biological processes, molecular functions, and cellular components in a species-independent approach. GO annotation is only available for identified novel genes and isoforms.
Genes were classified into one or several parts of GO by their functions. Relying on the GO annotation results, we could detect gene functions.
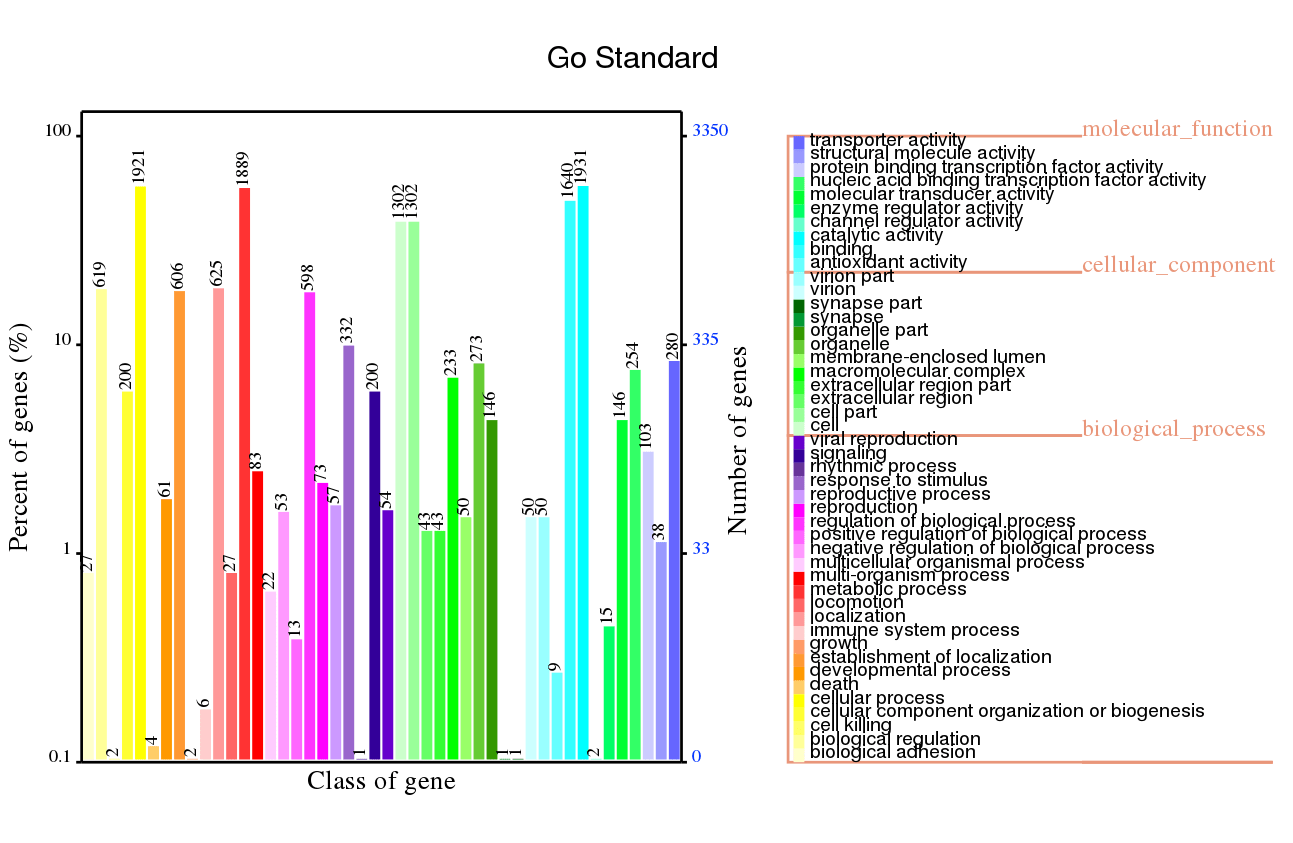
Note:
The horizontal axis displays the GO function class for the annotated genes, the right vertical axis is the gene number, and the left vertical axis is the percent of gene number annotated in all the coding genes.
KEGG Annotation
KEGG stands for Kyoto Encyclopedia of Genes and Genomes. It is a database resource for understanding high-end functionalities and utilities of the biological system, such as the cell, the organism, and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies. Using KEGG annotation, we could find genes that related to the annotated gene conveniently.
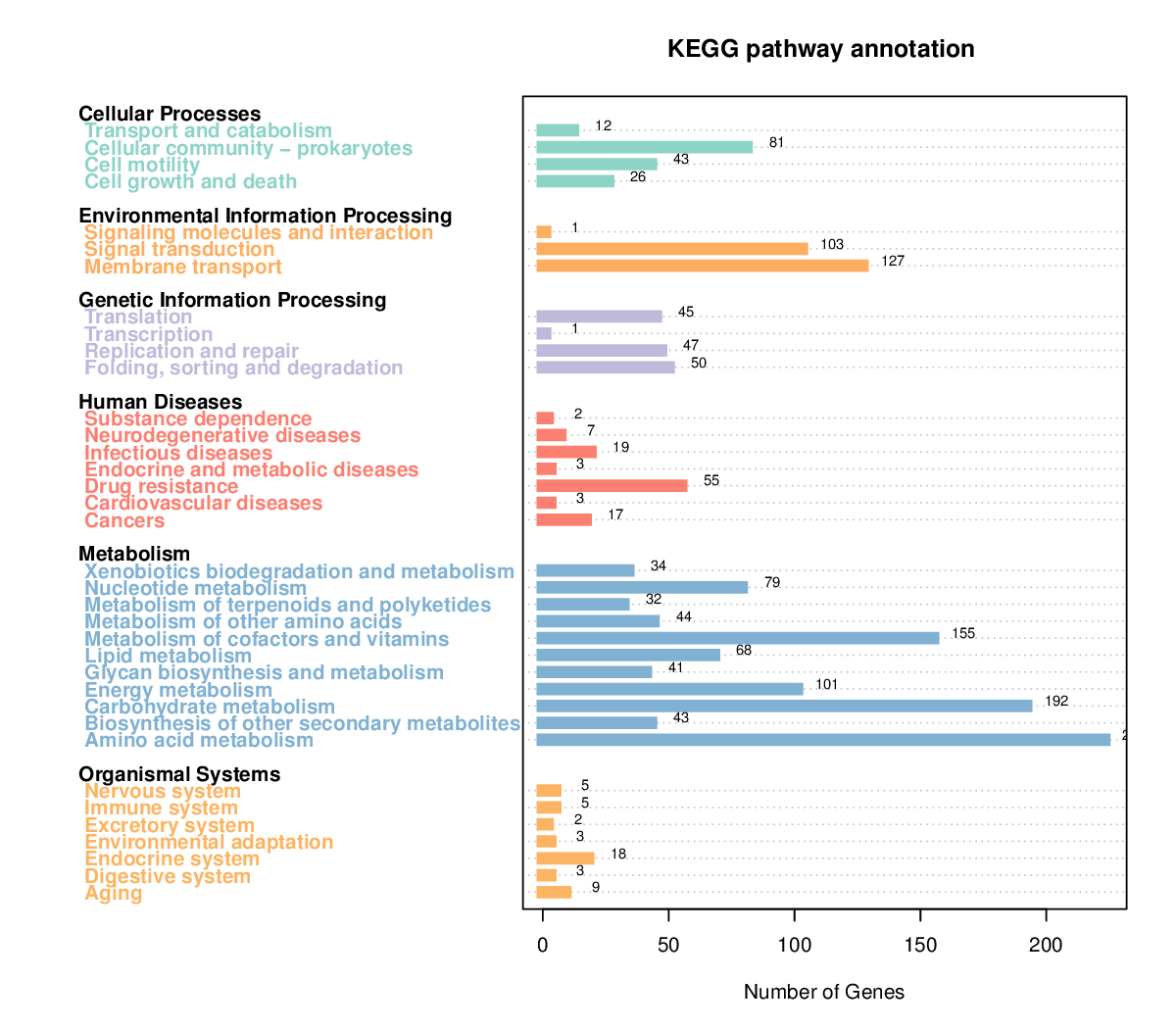
Note;
The horizontal axis is the KEGG pathway type, and the vertical axis shows the number of annotated genes.
COG Annotation
COG is the abbreviation of Cluster of Orthologous Groups of proteins. It is a protein database created and maintained by NCBI and is based on the evolution relation of the protein systems among bacteria, algae, and eukaryotes. COG annotations can be normally used to determine protein families. All proteins classified in one COG part contain homologous sequences that can be used to deduce the function of a protein.
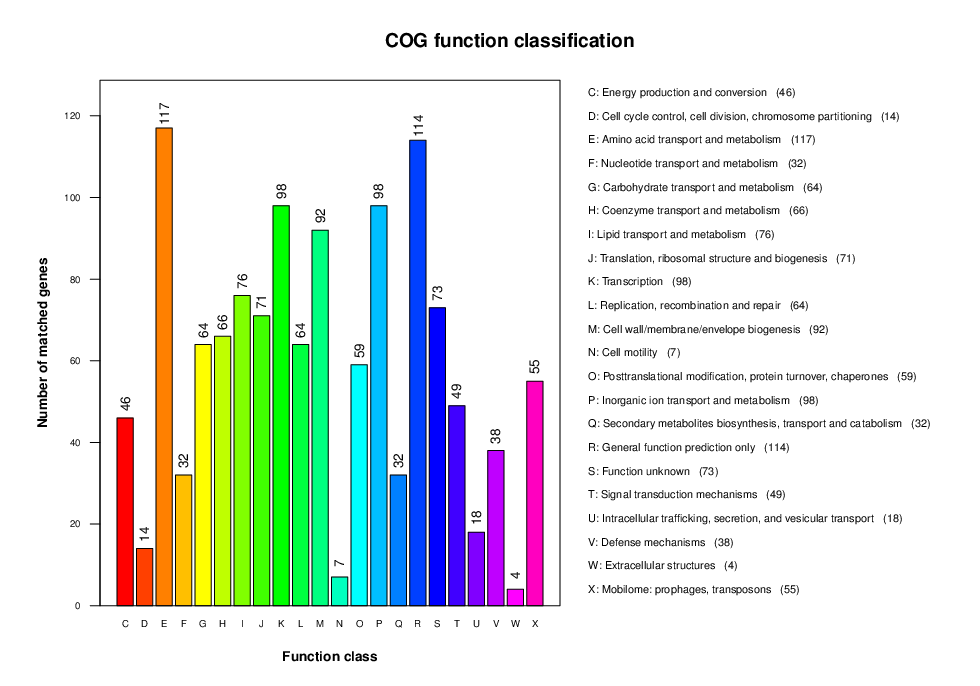
Note:
The horizontal axis is the COG function type, and the vertical axis is the number of annotated genes.
NR Annotation
The full name of NR is the Non-Redundant Protein Database. It is a protein database without duplication which is created and maintained by NCBI. The database is complete and the annotation results contain species information that can be used to classify different species.
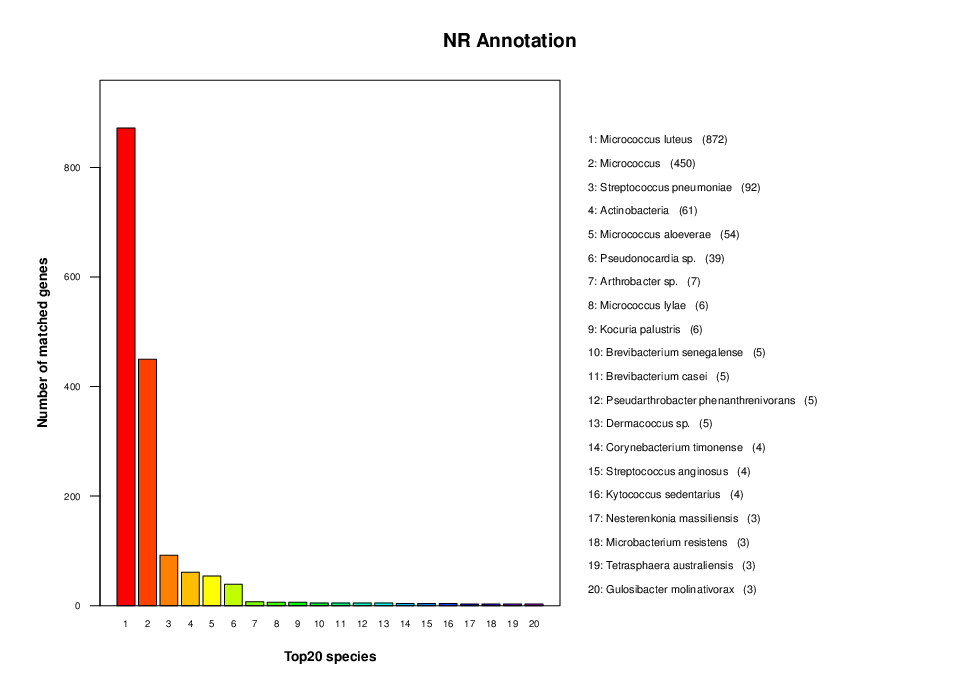
Note:
The horizontal axis is species ID, and the vertical axis is the number of annotated genes.
Short Read Sequencing (aiming at Draft Map and Survey)
Assembly Result
Sequenced reads were mapped to the assembly results, and then depth and GC content of assembly results were counted to further reflect the distribution of both GC content and the sequencing depth.
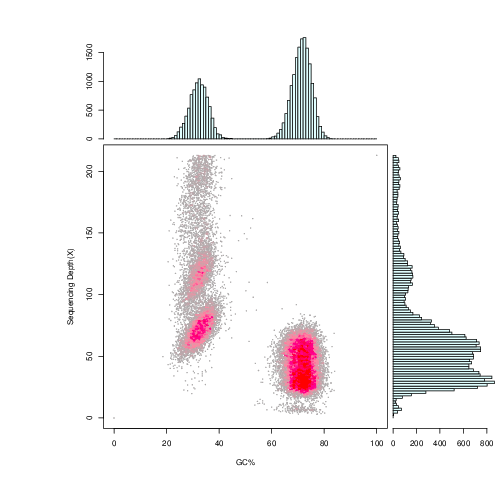
Note:
X-coordinate is GC content, and Y-coordinate is average depth. The right side is the distribution of sequence depth and the upper side is the distribution of GC content.
Gene length distribution
Generally, the microbial genome contains profuse functional regions, which account for even more than 90% of its size. In addition to the coding regions, non-coding regions are more likely to affect the regulation function of transcription, post-transcriptional modifications, translation, and epigenetic modifications. Part of the functional regions is related to the diversity of microbial evolution.
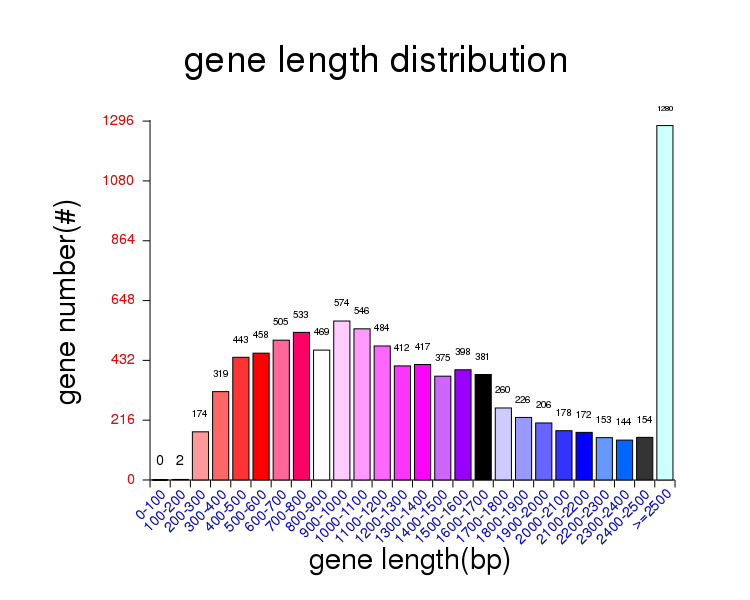
Note:
The constitution of the sequenced genome was learned using various methods, such as gene prediction, repeats prediction and non-coding RNA prediction, etc. X-coordinate is gene length and Y-coordinate is gene numbers.
*Please contact us to get full demo report.