Introduction to RNA Immunoprecipitation Sequencing (RIP-seq)

RNA immunoprecipitation sequencing (RIP-seq) is a high-throughput RNA sequencing method widely used to study Protein-RNA interactions to detect RNA interactions with the target proteins. The coupling of RIP-seq with other sequencing technologies enables the detection of a large number of RNA-protein complexes in a single run. RIP-seq provides deep insights not only into the central dogma (i.e., transcription and translation) but also in recent research areas like gene regulation by ncRNAs and RNA interference.
Applications of RNA Immunoprecipitation Sequencing (RIP-seq)
- To verify the interactions between RNAs and target proteins
- To identify genome-wide networks of RNA-RBP (RNA-binding protein) interactions
- To analyze the interactions between RBP and non-coding RNA (ncRNA), long non-coding RNA (lncRNA), and micro RNA (miRNA)
Benefits of RNA Immunoprecipitation Sequencing (RIP-seq)
- RIP-seq is multifaceted in interpreting the interaction network between RNA-RBP with respect to ncRNAs, such as lncRNA and miRNA.
- In comparison to CLIP-seq, RIP-seq does not need to perform UV-crosslinking which makes it easily operatable and warrant accurate results.
- The wide coverage allows screening and identification of protein binding sites accurately on the whole genome.
- Combining high-resolution and high-throughput technology enables us to find more new protein binding sites.
Latest Blogs
RIP-seq Specifications: RNA Sample Requirements
| Sample Type | Required Amount | Peaks Distribution | Purity |
| Enriched RNA Sample | ≥ 20ng (≥ 100ng for rRNA depletion) |
For unfragmented sample, fragment should be ≥ 1000 bp. For fragmented sample, fragment should be ≥ 80 bp. |
A260/280>2.0 |
RIP-seq Specifications: Sequencing and Analysis
| Sequencing Platform | Illumina NovaSeq 6000 Sequencing System |
| Read Length | Paired-end 150bp |
| Recommended Data Amount | ≥ 20 million read pairs per sample for the species with a reference genome |
| Content of Data Analysis |
|
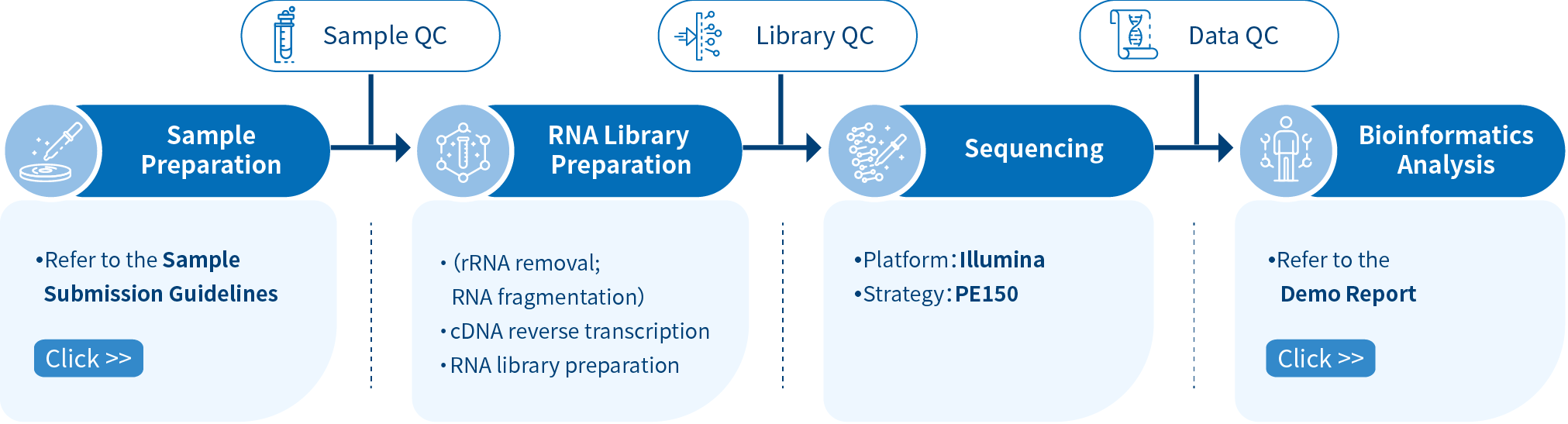
Novogene Workflow of RIP-seq Service
Novogene RIP-seq service comprises four steps, the first step being the sample preparation followed by RNA library preparation and Illumina PE150 sequencing, and finally, data analysis using bioinformatics pipelines. From RNA sampling to obtaining data reports, each step can influence the quality and quantity of data output, directly affecting the results of subsequent bioinformatics analysis. Novogene ensures stringent verification of each step, including sample quality control, library quality control, and sequencing data quality control, to ensure the high quality, accuracy, and reliability of sequencing data and provides comprehensive bioinformatics analysis.
Featured Publications using Novogene RIP-seq Service
-
Nature CommunicationsIssue Date: 21 March 2024IF: 14.7DOI: 10.1038/s41467-024-46754-2
-
YTHDF2 governs muscle size through a targeted modulation of proteostasis
Nature CommunicationsIssue Date: 11 March 2024IF: 14.7DOI: 10.1038/s41467-024-46546-8
-
ImmunityIssue Date: 28 February 2024IF: 32.4DOI: 10.1016/j.immuni.2024.02.002
-
Cancer DiscoveryIssue Date: 8 January 2024IF: 28.2DOI: 10.1158/2159-8290.CD-23-0486
-
Intrinsic targeting of host RNA by Cas13 constrains its utility
Nature Biomedical EngineeringIssue Date: 23 October 2023IF: 28.1DOI: DOI:10.1038/s41551-023-01109-y
Genome-wide Distribution of Peaks
The number of peaks mapping to the chromosome and its distribution reflect the distribution of the protein binding sites.
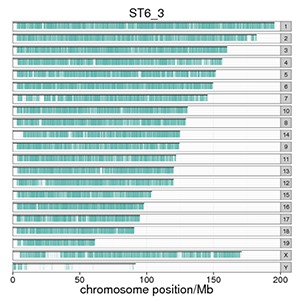
Note:
The X-axis is the number of peaks in the chromosome. The Y-axis shows the chromosome numbers. Every blue line represents a peak.
Motif Analysis
Systematic motif analysis is extremely encouraging and promising to comprehend the novel mechanisms in RNA trafficking and sorting as well as in other RNA applications. Motif, or conserved sequence, is shared by multiple binding sites in RNA.
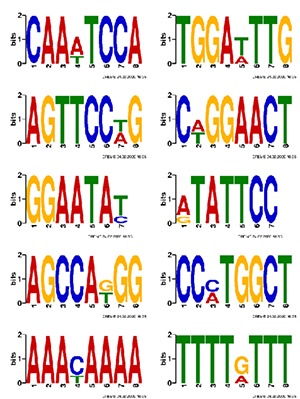
Note:
The figure uses a sequence logo to illustrate the base bias in different positions in the binding sites of a Motif.
Peak Distribution in the Functional Gene Regions
Distribution of peaks in the different functional areas.
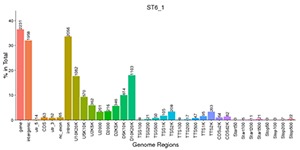
Note:
The X-axis represents different functional areas, and Y-axis represents the ratio of the peak in the functional region to the total peaks. The number on the top of the functional region represents the peak number.
*Please contact us to get the full demo report.