Introduction to Reduced Representation Bisulfite Sequencing (RRBS)

Reduced Representation Bisulfite Sequencing (RRBS) detects millions of novel CpG sites based on the improved bisulfite sequencing method, providing coverage to approximately all gene promoters, CpG islands, gene bodies, repetitive DNA sequences, and regulatory elements. It is an NGS-based sequencing platform and combines bisulfite sequencing and restriction enzymes to analyze the genome regions containing high CpG content. This combination allows reduced representation bisulfite sequencing to enhance the efficacy of the sample utilization and provides a perfect platform for clinical applications and pilot research.
By merging the NGS technology, bisulfite sequencing, and restriction enzymes, Novogene provides genome-wide methylation profiles at very affordable costs.
Applications of Reduced Representation Bisulfite Sequencing
- Applications in clinical research fields, such as tumor-subtype classification, molecular marker identification, drug target location determination, and pathological mechanism studies
- Applications in crop development, crop adaptability, and agronomic traits in the agricultural sector
Benefits of Reduced Representation Bisulfite Sequencing
- Cost-effective relative to WGBS in mammalian research.
- Precise localization of methylation sites with single-base resolution.
- Covers millions of CpG sites genome-wide.
- Low input DNA requirement.
RRBS Specifications: DNA Sample Requirements
| Sample Type | Required amount | Purity |
| Genomic DNA | ≥ 800 ng | A260/280=1.8-2.0 |
RRBS Specifications: Sequencing and Analysis
| Sequencing Platform | Illumina NovaSeq 6000 Sequencing System |
| Read Length | Paired-end 150 bp |
| Recommended Data Amount | ≥ 10 Gb clean data per sample |
| Content of Data Analysis |
|
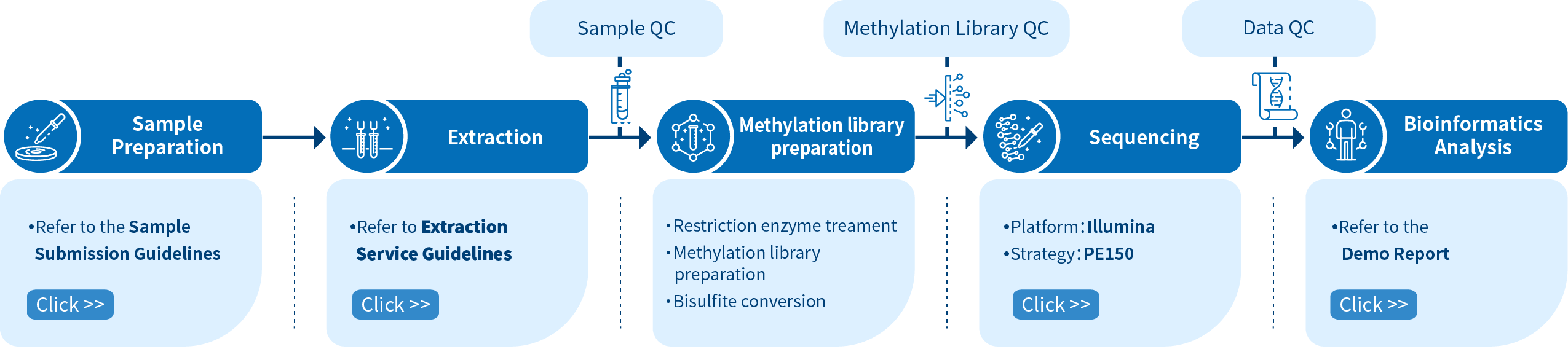
Project Workflow of Novogene RRBS Service
The Novogene RRBS service comprises four steps. The first step involves sample preparation which is followed by library preparation, sequencing, and finally data analysis using bioinformatics pipelines. To construct a methylation library, RNA fragments of various lengths are created using a restriction enzyme treatment, followed by sodium bisulfite conversion of unmethylated cytosines into uracils. Libraries are then sequenced using Illumina PE150 and eventually data extraction and bioinformatics analysis are performed.
Novogene audits every experimental step strictly to ensure the accuracy and reliability of the sequencing data. The quality control of the RRBS library pool is performed to ensure high-quality data output fundamentally. Obtaining good-quality data is the premise to ensure that bioinformatics analysis is correct, comprehensive, and credible.
Featured Publications using Novogene RRBS Service
-
BMC Plant BiologyIssue Date: 6 June 2024IF: 4.3DOI: 10.1186/s12870-024-05197-z
-
Aberrant DNA methylation distorts developmental trajectories in atypical teratoid/rhabdoid tumors
Life Science AllianceIssue Date: 18 March 2024IF: 4.4DOI: 10.26508/lsa.202302088
-
The RNA m5C modification in R-loops as an off switch of Alt-NHEJ
Nature CommunicationsIssue Date: 30 September 2023IF: 16.6DOI: 10.1038/s41467-023-41790-w
-
A fast chemical reprogramming system promotes cell identity transition through a diapause-like state
Nature Cell BiologyIssue Date: 7 August 2023IF: 21.3DOI: 10.1038/s41556-023-01193-x
-
An atlas of regulatory elements in chicken: A resource for chicken genetics and genomics
Science AdvancesIssue Date: 3 May 2023IF: 14.957DOI: 10.1126/sciadv.ade1204
Methylation Level Distribution in Whole Genome
The whole genome is divided into sub-bins by 10kb. We analyze the C sites in every context for each sample (or each group, combine the samples if the biological replicates are > 5).
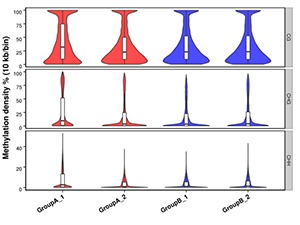
Note:
Methylation density for each comparison group.
Circos Plots for Methylation Density on Chromosome
Circos plots are typically used to demonstrate massive genomic data across several chromosomes within a single plot. We have used Circos plot to represent the methylation density on a chromosome (Silin Zhong , James J Giovannoni et al. (2013) and Krzywinski , M. et al. (2009) Circos )
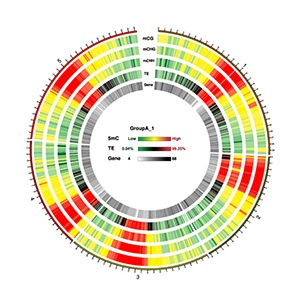
Note:
Circos plot represents (from outside to inside):1. The heatmap for methylation density in CG context 2. The heatmap for methylation density in CHG context 3. The heatmap for methylation density in CHH context 4. The heatmap for TE(repeat elements)rate 5. The heatmap for gene density
Methylation Level Distribution of All Samples in Genomic Features
The average methylation levels with different cytosine contexts are calculated in different functional genomic regions such as promoter, exon, intron, CGI, CGI shore, repeat(Promoter region is the 2kb region above TTS, 5’UTR, Exon and Intron are obtained from the Ensembl structure annotation files).
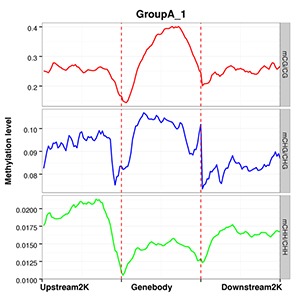
Note:
The x-axis represents different genomic elements, and the y-axis represents methylation levels. The various functional areas of each gene are divided into 20 bins, and the average methylation level within each bin region is calculated. Different colours stand for different contexts (CpG, CHG, CHH).
Heatmap Analysis of Gene Functional Region Methylation Levels in Different Sequence Environments of Different Samples
The heatmap analysis of the methylation level of the functional regions between samples reflects the degree of agreement of the methylation level distribution of each sample in the gene function region. Data of this heatmap analysis is extracted from the methylation level of genomic features for a single sample.
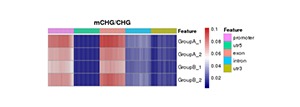
Note:
In the figure, darker red means high methylation levels, and darker blue depicts low methylation levels.
*Please contact us to get the full demo report.