What you can explore with non-coding RNA data
Many of the non-coding RNAs (ncRNAs) produced by eukaryotic cells have been demonstrated to have crucial roles in biological processes. ncRNAs do not have the potential to be translated into proteins. They actively participate in the regulation, transcription, or post-transcriptional changes of gene expression. The presence, quality, and function of ncRNA in the gene expression of a biological sample at a specific time are revealed by RNA sequencing using next-generation sequencing technology. The ncRNAs also serve the role of reliable biomarkers for diagnoses and have functions in epigenetics.
Long non-coding RNAs (lncRNAs), short non-coding RNAs (sRNAs), and circular non-coding RNA (circRNA) are the three different forms of ncRNAs. Long non-coding RNAs have lengths of more than 200 nucleotides. They play their role to modify chromatin structure and function, and the transcription of nearby and distant genes. The sRNAs are about 22 nucleotides in length with a range of 15-32 and are involved in gene silencing and posttranscriptional regulation of gene expression by binding to messenger RNAs. One of the most stable types of non-coding RNAs, circular RNAs are single-stranded RNA molecules and form a covalently closed continuous loop. Their stability and resistance to externally placed digestion give a great advantage during library generation for NGS. They have several functions; the most well-known function is acting as miRNA sponges and protein decoys. Other functions include transcription regulation and protein translation.
The bioinformatics analysis of ncRNA sequencing involves the following steps.
- Quality control
- Mapping
- Annotation
- Quantitative analysis (expression)
- Functional analysis
- Qualitative analysis (characterization)
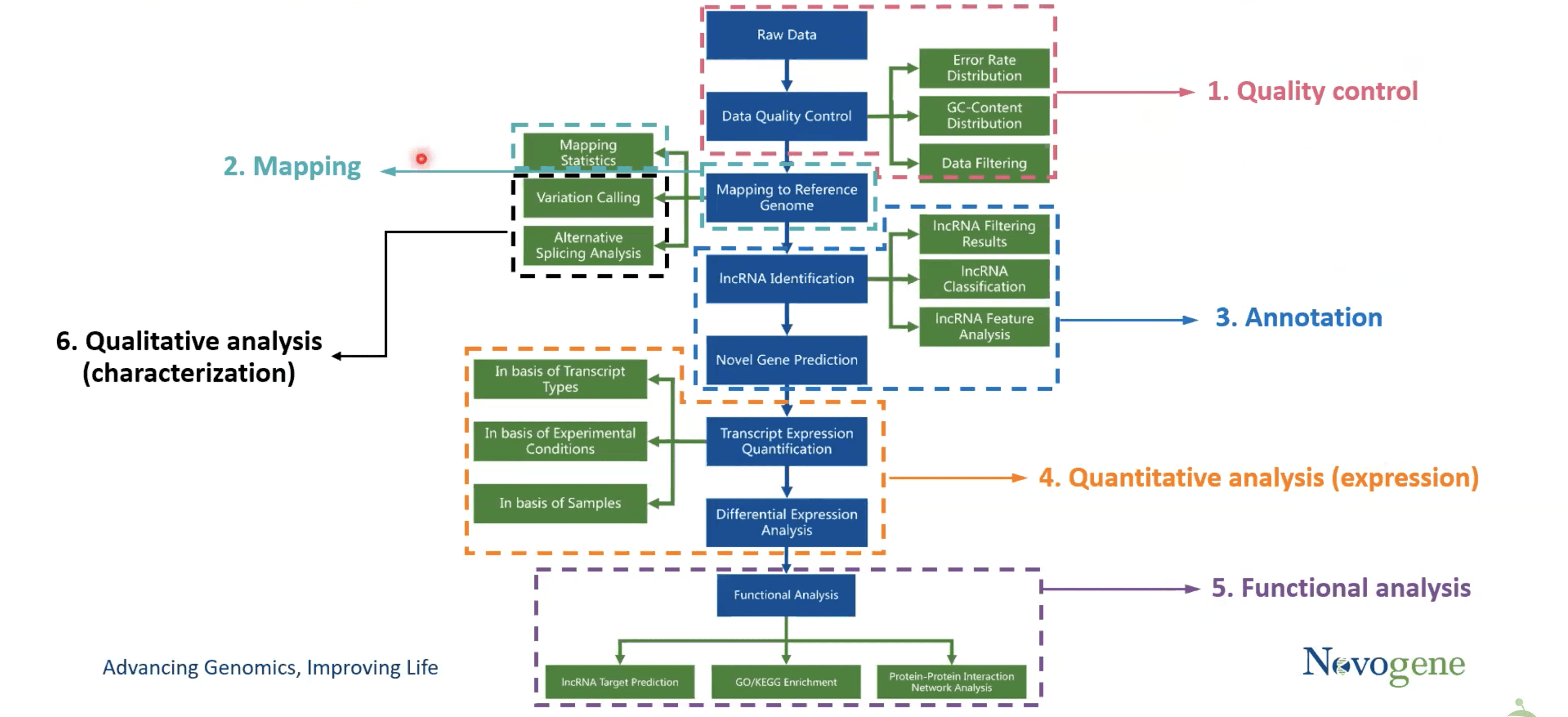
1.Quality control
Here, first, Raw data is generated and stored in FASTQ format (a text-based format for storing a nucleotide sequence). The FASTQ format has four different rows for “Sequence ID”, Read bases, separator, and quality score provider of the FASTQ format. Moving on to the data filtering step, which uses the fast program to introduce raw reads and produce clean reads. In the end, three things; error rate, base content, and the portion of raw reads transformed into clean reads, are attained.
2.Mapping
On the clean reads, mapping is performed using a program called Hierarchical Indexing for Spliced Alignment of Transcripts (HISAT2). The reference genome is indexed using a graph-based method, and the Bowtie2 algorithm is used for alignment. This method yields more accurate results with quick and sensitive alignment. Here, the output is a binary form of a SAM (Sequence Alignment Map) file called a BAM file. These BAM files may now be seen in the Integrative Genomics Viewer and compared to the reference genome to determine their differences.
3.Annotation
After getting BAM files, annotation is done through a software named StringTie. Annotation means identifying functional elements along the sequence of a genome. It uses a network for algorithm as well as an optional de novo assembly step to assemble steps into known or novel gene models based on known gene annotations. In this case, BAM files plus reference annotation files are introduced (input) and a GTF is obtained (output) through transcript annotation of the assembled and aligned reads. Now the assembled transcripts are merged to remove duplicate or redundant transcripts. After this, different filters (Exon number filter, transcript length filter, coding potential filter, etc.) are used to identify and predict the ncRNA types.
4.Quantitative analysis
The simplest approach to quantify the ncRNA and coding gene is to count the number of reads that map to each transcript. However, two factors need to be taken into consideration. First, the estimated expression level depends on the read counts and total reads sequenced for each sample and the second is that read counts also depend on total gene/transcript length. This means it is essential to perform a normalization step to make the data comparable between and within samples.
5.Functional analysis
In functional analysis, biological reference is assigned to a set of genes. It is determined whether there is the enrichment of any known biological functions, interactions, or pathways. So, a software called ClusterProfiler is used which implements methods to analyze and visualize functional profiles of genomic coordinates, gene and gene clusters and enrich the data. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) are the most frequently used databases for functional analysis. Aside from data enrichment, ncRNA target prediction can also be done.
6.Qualitative analysis
In this step, variant discovery, and alternative spicing (AS) is done through software; GATK and rMATS respectively. GATK is used to identify where the aligned reads differ from the reference genome and write to a variant call format (VCF) file. BAM files are introduced and VCF files are obtained. On the other hand, rMATS is designed for detecting differential AS in replicated RNA-seq data.
ncRNA interaction and regulatory networks
Competing Endogenous RNAs theory says that lncRNAs, mRNAs and circRNAs have a competition relationship, trying to interact with the miRNAs and depending on who interacts with which type of microRNA is going to change the gene expression and the transcription regulation. lncRNA and mRNA co-expression analysis can be done via Pearson analysis. LncRNA-mRNA interaction can be visualized through a network software “Cytoscape’.
Click here to watch the full video
How Novogene Can Help
Click here to learn more about our Non-coding RNA sequencing service
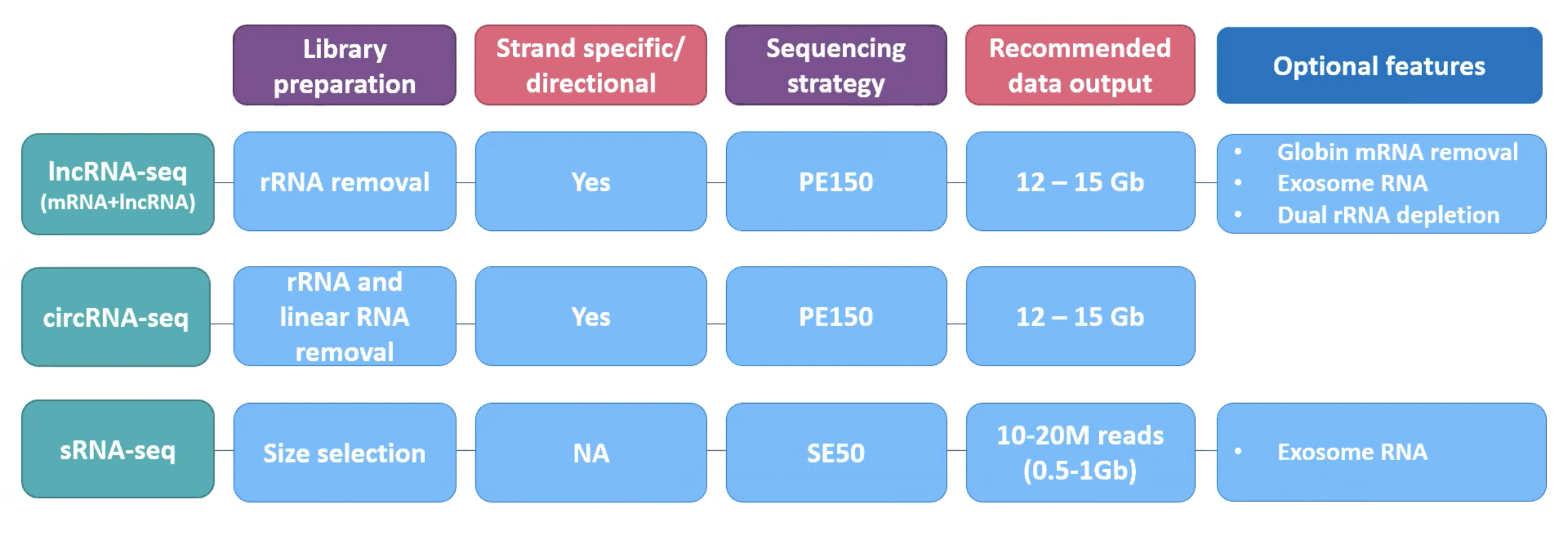
The company’s mission is to advance genomics and improve life. The services offered by Novogene are whole genome and transcriptome analysis, epigenome, human, microbial, plant, and animal genome sequencing, etc. The services cover a huge range of real-life applications from complex disease research, oncology, forensics, food, and water safety to biopharma and industrial diagnosis. The state-of-the-art lab, latest Novaseq 600 sequencing machines, Falcon II sequencing delivery platform and supercomputing capacity of Novogene provide faster results with reduced human error and time requirements. In the case of ncRNA sequencing, the Novogene can prepare rRNA removal libraries, with a sequencing strategy of PE150, keeping strand-specific directional library by default for lncRNA-seq and circRNA-seq. Optional features of Globin mRNA removal and exosome RNA are also provided. To avoid bacterial contamination dual rRNA depletion strategy is adopted. For sRNAs removal and directional libraries are not needed due to their small sizes and the SE50 strategy is adopted. Novogene also provides only sequencing services for premade libraries as well. Novogene will always provide more efficient & higher quality services in order to be your trusted partner in genomics.