Understanding Low-Pass Whole-Genome Sequencing: Benefits and Applications
What is Low-Pass Whole-Genome Sequencing (LP-WGS)?
In the ever-evolving field of genome analysis, researchers are continually seeking cost-effective and efficient methods for comprehensive sequencing. Low-Pass Whole Genome Sequencing (LP-WGS) offers a streamlined solution that balances affordability with extensive genomic coverage. LP-WGS is a high-throughput, shallow sequencing method that provides a broad view of the genome at a lower depth of 0.1x to 0.5x, making it a cost-effective alternative to traditional whole genome sequencing approaches.
LP-WGS not only reduces the costs associated with variant discovery but also delivers higher statistical power compared to genotyping arrays. Additionally, its reduced sample requirements make LP-WGS particularly valuable for addressing complex biological questions involving large-scale population genetics studies.
Key Advantages of Low-Pass Whole-Genome Sequencing
- Cost-Effectiveness: LP-WGS offers a significant reduction in per-sample sequencing costs, thus making it more cost-efficient for large-scale studies or clinical testing.
- Increased GWAS Power and Accuracy: LP-WGS provides a substantial increase in statistical power for genome-wide association studies (GWAS) and improves accuracy in polygenic risk prediction.
- Broader Variant Detection: LP-WGS can detect novel variants, structural variations like CNVs, and loss of heterozygosity across the entire genome, offering a more comprehensive view of genetic variation.
- Low DNA Input Requirements: LP-WGS requires substantially less DNA input compared to high-depth sequencing approaches. This feature of LP-WGS is particularly advantageous when working with limited or degraded, as it enables comprehensive genomic analysis with minimal DNA input.
- Rapid Turnaround Time: Due to its lower depth of coverage, LP-WGS offers quicker turnaround times compared to high-pass sequencing methods, making it a preferred choice for large-scale studies or time-sensitive research.
Bioinformatics Analysis and Tools for LP-WGS Data Analysis
For successful implementation of Low-pass or Shallow whole genome sequencing, a structured methodology is followed. Beginning with the careful preparation of biological samples, the process continues through sequencing and subsequent bioinformatics analysis. After sequencing, the LP-WGS data is subjected to various statistical computations and bioinformatics techniques for customized analysis, accurate interpretation and gaining robust genetic insights even with lower sequencing depths (Figure 1).
Some of the key steps and tools utilized for subsequent analysis are listed below.
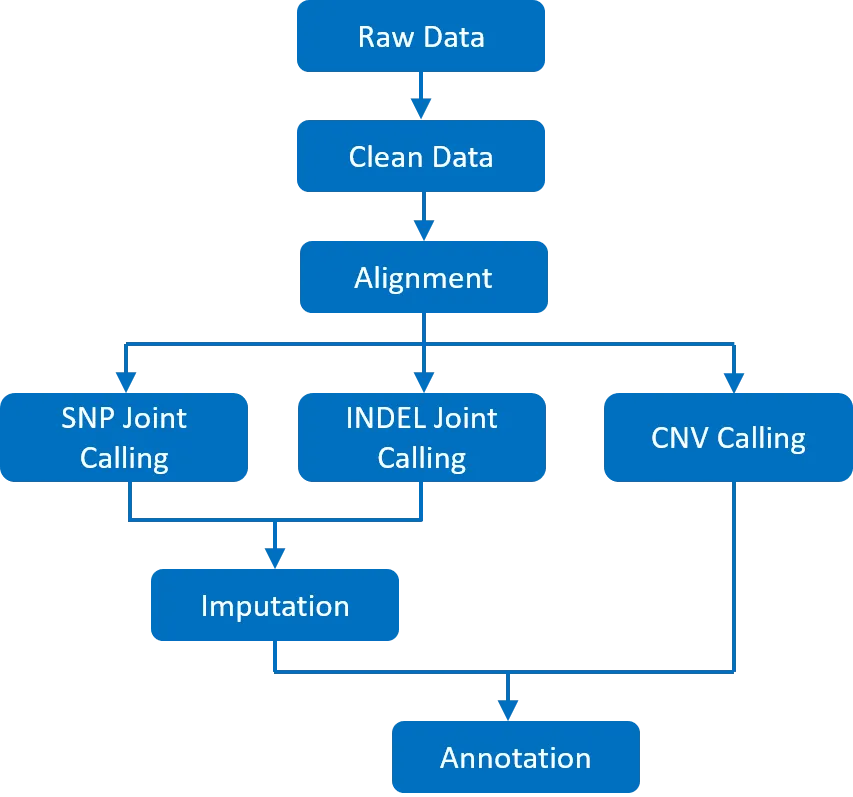
Figure 1: Bioinformatics Workflow of LP-WGS Data
- Quality Control: Quality control checks are essential to assess the accuracy and completeness of the sequencing data. Tools such as BEDTools and Picard are used to evaluate coverage, detect potential biases, and ensure data integrity.
- Read Alignment: After obtaining clean data, the first step is aligning the raw sequencing reads to a reference genome. Tools such as BWA (Burrows-Wheeler Aligner) and Bowtie2 are commonly used for this purpose. Accurate alignment is crucial for identifying genetic variants and ensuring that the sequencing data is mapped correctly across the genome.
- Variant Calling: Aligned reads undergo variant calling to identify genetic variants such as single nucleotide polymorphisms (SNPs) and insertions/deletions (Indels). Tools like GATK (Genome Analysis Toolkit) and SAMtools are widely used for this process. These tools help in detecting variations even at low coverage depths typical of LP-WGS.
- Imputation: In cases where coverage is sparse, imputation tools such as Minimac and IMPUTE can be employed to infer missing genotype. Application of imputation algorithms improves the completeness of the data, allowing up to 99% accurate variant detection.
- Variant Annotation: ANNOVAR is commonly employed for variant annotation, highlighting changes in protein-coding regions and affected genomic areas. Databases like RefSeq and Gencode help identify genomic regions and potential protein alterations. For allele frequency, databases such as 1000 Genomes, ExAC, and gnomAD are utilized. Predictive tools like SIFT, PolyPhen, and CADD assess the deleterious impact of mutations, while conservation scores from GERP++ and phyloP determine the evolutionary importance of sites. Functional annotations are provided by databases like KEGG, Reactome, and Gene Ontology.
Case study:Comparative Analysis of Low-Pass Sequencing and Imputation in Companion Animals
In a recent study titled “A Cautionary Tale of Low-Pass Sequencing and Imputation with Respect to Haplotype Accuracy” researchers from the Roslin Institute and the Royal (Dick) School of Veterinary Studies at the University of Edinburgh delve into the effectiveness of low-pass sequencing and imputation in terms of haplotype accuracy in companion animals.
The researchers isolated DNA from the saliva of 30 Labrador Retrievers. Library preparation (150 bp paired-end, 350 bp insert size) and whole genome sequencing (NovaSeq 6000) were performed by Novogene (UK). The DNA samples were sequenced at various depths: low (0.9X and 3.8X), high (43.5X), and down-sampled from 43.5X to 9.6X and 17.4X. They also compared the performance of genotype imputation using a diverse reference panel of 1021 dogs and two subsets of this panel, each containing 256 dogs with varying breed compositions.

The paper assesses the concordance of imputed genotypes across different sequencing depths and reference panels, noting that larger panels with greater allelic diversity tend to reduce imputation errors. Thus, rare variants with a minor allele frequency (MAF) of 0.01 or less proved challenging to impute accurately.
The study also examines the correlation of association analysis results between different sequencing depths using single-marker (GEMMA) and haplotype-based (XP-EHH) tests. While single nucleotide variants (SNVs) linked to the chocolate coat phenotype were successfully validated using the GEMMA method, the haplotype-based XP-EHH method showed lower sensitivity. This was surprising given the extensive linkage disequilibrium and large haplotype blocks in pedigree dogs.
Though the potential of low-pass sequencing and imputation, especially in livestock breeding programs have been intensively proved. Those findings suggest the need for caution when using low-pass sequencing and imputation, particularly in ensuring accurate haplotype phasing and genotype concordance in companion animal genomics. Future studies could mitigate this by incorporating more comprehensive reference panels and advanced phasing techniques. As technology advances, LP-WGS is poised to play a pivotal role in uncovering new insights across the field of omics.
More Applications of Low-Pass Whole-Genome Sequencing in Human
Low-Pass Whole Genome Sequencing (LP-WGS) has notable applications across research, clinical diagnostics, and population studies.
- Genome-Wide Association Studies (GWAS): LP-WGS improves GWAS by identifying both common and rare genetic variants, enhancing the study’s statistical power and accuracy in detecting genetic associations.
- Prenatal Testing: Used in non-invasive prenatal testing (NIPT), LP-WGS detects chromosomal abnormalities like trisomies from fetal DNA in maternal blood, offering a reliable and low-risk alternative to traditional tests.
- Evolutionary and Ancestry Analysis: LP-WGS helps study human evolution and population genetics by identifying genetic markers that reveal ancestral relationships and migration patterns.
- Pharmacogenomics: LP-WGS aids in identifying genetic variants that affect drug metabolism and efficacy, helping to personalize medication plans based on genetic profiles.
About Novogene
Novogene is one of the first few companies in the world employing the powerful platform, capable of sequencing up to 280,000 human genomes per year at the lowest cost per genome. We have extensive experience providing both shallow or deep whole genome sequencing service on this powerful system with high quality results. With the throughput and capacity of the Illumina platform, Novogene’s deep experience with the system, and our advanced bioinformatics capabilities, our company is able to expertly meet customer needs for executing large projects with timely turn-around and the highest quality results. Contact us directly, please go here.
References
https://www.sciencedirect.com/science/article/pii/S152515782030297X
https://pubmed.ncbi.nlm.nih.gov/37807935/
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8015847/
https://academic.oup.com/clinchem/article/69/5/510/7030091
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC7999180/
https://academic.oup.com/g3journal/article/14/2/jkad276/7457298
https://pubmed.ncbi.nlm.nih.gov/37355983/
https://gsejournal.biomedcentral.com/articles/10.1186/s12711-024-00875-w