WGS vs WES: Which Genetic Sequencing Method is Right for You?
Whole genome sequencing (WGS) and whole exome sequencing (WES) are two of the most popular methods used in modern genetic research. While both methods provide a comprehensive view of an individual’s genetic makeup, there are some key differences between the two techniques that researchers need to consider when choosing which method to use. In this article, we will explore the differences between WGS and WES and their relative advantages and disadvantages. The first involves sequencing the entire genome, including both the protein-coding and non-coding regions, which enables researchers to identify almost all the changes that have occurred in a patient’s DNA. WES on the other hand only examines the protein-coding regions. Both methods have their own strengths and limitations and have broad applications in genomics research.
Advantages and Disadvantages of WES
One of the major advantages of WES is that it is a cost-effective way to sequence a large number of samples. Since only the exome is sequenced, the amount of data generated is significantly less than WGS, which can result in lower sequencing and analysis costs. Additionally, since the exome contains the majority of known disease-causing variants, WES is a powerful tool for identifying genetic causes of disease.
 Fig 1. WES Specifications: Sequencing and Analysis
Fig 1. WES Specifications: Sequencing and Analysis
The exome or protein-coding region of the genome accounts for less than 2% of the genome but is where most of the genetic variants associated with human disease are found. Research has shown that WES can be a useful tool for advancing clinical diagnostics. For example, Retter et al (2016) analyzed a large consecutive series of 3040 clinical WES cases and found that WES had an overall diagnostic yield of 28.8%2. More specifically, the diagnostic yield was 23.6% for proband-only cases, increasing to 31% when three family members were analyzed. WES also has the potential as a tool for the earlier diagnosis of genetic diseases. Delanne et al (2021) examined WES as a tool to diagnose inherited metabolic disorders (IMD) in 547 patients who presented with unspecified developmental disorders. WES identified 32% of this cohort as having a positive diagnosis, 12% of which were diagnosed with 15 different IMDs. Remarkably, this study also confirmed that WES can be used to diagnose IMDs in fetuses with unspecified symptoms.
Finally, WES can also be used to advance cancer research, enabling researchers to identify variants or mutations that play a role in tumor progression. WES offers comprehensive coverage of coding regions and a depth of coverage that can be used to identify both common and rare variants.
However, one of the disadvantages of WES is that it may miss important variants that are outside of the exome. This can be particularly problematic for variants located in regulatory regions, which are critical for gene expression and regulation. WES is also not able to identify structural variants or large insertions and deletions.
Advantages and Disadvantages of WGS
One of the major advantages of WGS is that it provides a more comprehensive view of an individual’s genetic makeup. WGS can identify variants that are not present in the exome, including those in non-coding regions and structural variants. This can be particularly useful for identifying rare or novel variants that may be missed by WES. Additionally, WGS can provide information about ancestry and population genetics.
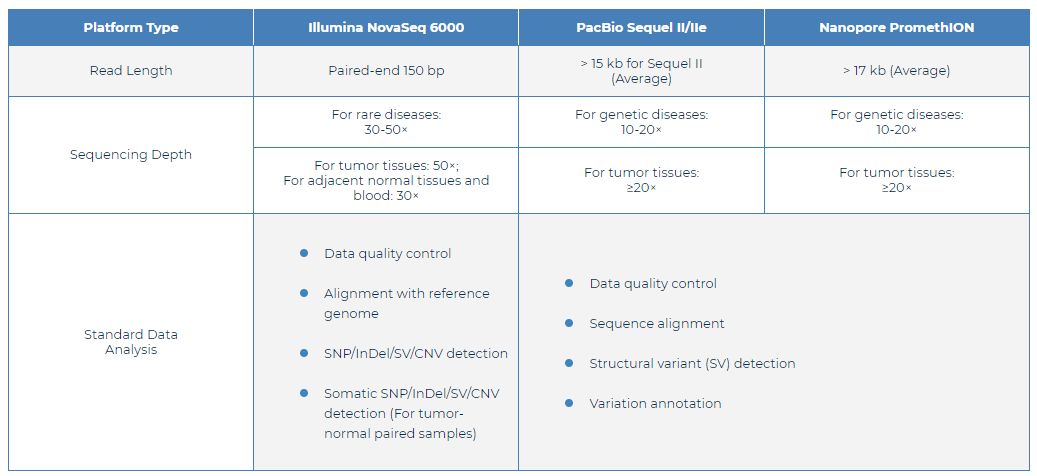 Fig 2. WGS Specifications: Sequencing and Analysis
Fig 2. WGS Specifications: Sequencing and Analysis
WGS is supporting a new era of personalized medicine, whereby researchers can sequence the entire genome of a patient and examine their genome for genetic variations. WGS is one of the most widely used applications, providing vast amounts of data the world over. As of 2015, it was reported that 2504 genomes of people from 26 different populations had been reconstructed. Further, as of the same year, there were 99 mammalian reference genes available on Genebank and 57 of these had used only Illumina technology, while a further 16 combined Illumina sequencing with Sanger sequencing .
The increase in the use of WGS technologies has provided more comprehensive information about a patient’s genetic material than traditional methods that have focused largely on variations in single nucleotide polymorphisms (SNPs), omitting other forms of variation such as copy number variants (CNVs). CNVs can occur in both coding and non-coding regions and approximately 18% of the variation in the expression level of around 15,000 genes can be attributed to copy number difference8. In addition, CNVs have been linked to a range of complex diseases such as Parkinson’s disease, Alzheimer’s disease, and chronic pancreatitis .
However, one of the major disadvantages of WGS is that it is more expensive than WES due to the larger amount of data generated. WGS also requires more computational resources and expertise to analyze the data. Additionally, WGS may generate more false positive results, particularly for low-frequency variants, which can complicate the interpretation of results.
Which Method is Right for Your Research?
The choice between WES and WGS depends on the research question and available resources. If the research question is focused on identifying disease-causing variants in protein-coding regions, then WES may be the most appropriate method. It costs a lot less than WGS as it is used to target less than 2% of the genome. This is useful if you are targeting a known gene or set of genes, or a disease associated with coding regions as it allows you to increase the sample number. This is also important for large population comparisons. In addition, WGS data is an order of magnitude larger than WES data which can make interpretation more challenging.
If the research question is broader and requires a more comprehensive view of an individual’s genetic makeup, then WGS may be more appropriate. It allows you to examine a lot more of the genome than WES, and examine single-nucleotide variants, indels, structural variants, and CNVs in both the coding and non-coding parts of the genome. This can be especially important when examining diseases that are linked to non-coding regions of the genome. WGS also has more reliable sequence coverage and coverage uniformity. Differences in the hybridization efficiency of the capture probes used in WES can result in little or no coverage in certain areas of the genome.
In conclusion, WGS offers more comprehensive coverage than WES by allowing you to examine the genome as a whole for variations that could be linked to diseases. Fortunately! Recent advancements in instrumentation sequencing technology have made it more accessible than ever to conduct large-scale studies in a fraction of the time it once took. Thanks to increased usage and a reduction in cost, the possibilities for whole genome sequencing (WGS) are now limitless. With these new tools, researchers can dive even deeper into the mysteries of genetics and unlock groundbreaking discoveries that were once thought impossible. So, get ready to accelerate your research and push the boundaries of what’s possible with WGS. The future of genomics is here!
References
- Resta, C. D., Galbiati, S., Carrera, P., & Ferrari, M. (2018). Next-generation sequencing approach for the diagnosis of human diseases: Open challenges and new opportunities. EJIFCC, 29(1), 4-14.
- Retterer, K., Juusola, J., Cho, M.T., Vitazka, P., Millan, F., Gibellini, F., Vertino-Bell, A., Smaoui, N., Neidich, J., Monaghan, K.G. and McKnight, D., 2016. Clinical application of whole-exome sequencing across clinical indications. Genetics in Medicine, 18(7), pp.696-704.
- Delanne, J., Bruel, A.L., Huet, F., Moutton, S., Nambot, S., Grisval, M., Houcinat, N., Kuentz, P., Sorlin, A., Callier, P. and Jean-Marcais, N., 2021. The diagnostic rate of inherited metabolic disorders by exome sequencing in a cohort of 547 individuals with developmental disorders. Molecular Genetics and Metabolism Reports, 29, p.100812.
- 4.1000 Genomes Project Consortium, 2015. A global reference for human genetic variation. Nature, 526(7571), p.68.
- Walter, K., Min, J.L., Huang, J., Crooks, L., Memari, Y., McCarthy, S., Perry, J.R.B., Xu, C., Futema, M., Lawson, D. and Iotchkova, V., 2015. Management committee (2015). the uk10k project identifies rare variants in health and disease. Nature, 526(7571), pp.82-90.
- 6.1000 Genomes Project Consortium, 2010. A map of human genome variation from population scale sequencing. Nature, 467(7319), p.1061. 1000 Genomes Project Consortium, 2010. A map of human genome variation from population scale sequencing. Nature, 467(7319), p.1061.
- Estivill, X. and Armengol, L., 2007. Copy number variants and common disorders: filling the gaps and exploring complexity in genome-wide association studies. PLoS genetics, 3(10), p.e190.
- Redon, R., Ishikawa, S., Fitch, K.R., Feuk, L., Perry, G.H., Andrews, T.D., Fiegler, H., Shapero, M.H., Carson, A.R., Chen, W. and Cho, E.K., 2006. Global variation in copy number in the human genome. nature, 444(7118), pp.444-454.
- Chaisson, M., Wilson, R. & Eichler, E. Genetic variation and the de novo assembly of human genomes. Nat Rev Genet 16, 627–640 (2015).https://doi.org/10.1038/nrg3933