Uncovering the Genetic Basis of Rare and Complex Diseases through Whole Genome Sequencing (WGS)
As the cost of sequencing decreases and more efficient sequencing technologies are developed, high throughput sequencing methods are powerful tools for investigating and diagnosing complex diseases. Whole-genome sequencing (WGS), for example, is not only used as a primary tool for diagnosing rare diseases,[1] but may also become common for detecting repeat amplification diseases. This is paving the way for a new era of medicine that offers hope to patients and their families affected by rare and complex diseases.
Keywords: WGS, rare and complex diseases, GWAS, neurological disorders
Sequencing and the Role in Understanding Complex Diseases
The locus-specific nature of standard genetic testing methods may result in underdiagnosis, especially in pediatric patients, Ibañez et al. evaluated WGS as a diagnostic tool for neurological repeat expansion disorders.[1] First, the diagnostic accuracy of WGS detection of common repeat expansion loci associated with neurological outcomes were investigated. Samples from 404 patients suspected of having neurological disorders who had previously received PCR testing for repeat expansions were sequenced. Compared to the standard PCR test results, WGS (plus Expansion Hunter and visualization of expanded alleles) showed 97.3% sensitivity and 99.6% specificity. After that, a total of 11,631 undiagnosed patients with a suspected neurological disorder were tested to investigate whether WGS detection of repeat expansion could resolve diagnoses. Overall, repeat expansions were detected and visually confirmed in 105 patient samples, of which 81 were available for confirmatory PCR testing. Repeat expansions were PCR-confirmed in 68 of the 81 samples, and 13 were not confirmed (16% false discovery rate). Altogether, these findings support the utility of WGS as a clinical tool for identifying repeat expansion loci.
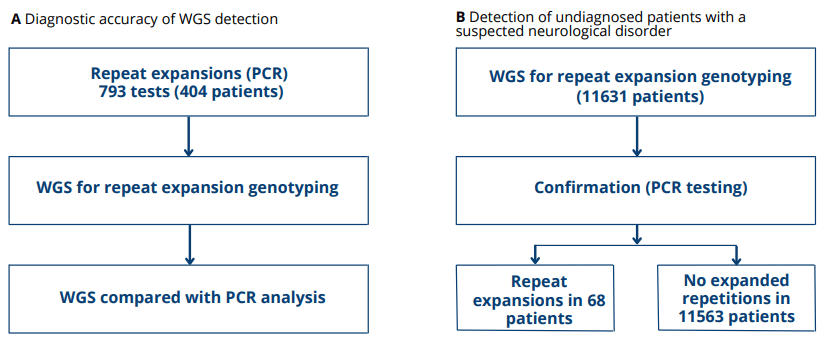
Figure 1 (A) WGS for the purpose of repeat expansion detection. (B) Validation in individuals who had a neurological condition that was hypothesized but went undetected by earlier genetic testing.
WGS is not the only sequencing technology relevant to complex diseases. In another example, Han et al. applied transcriptional profiling to reveal potential mechanisms of neuropsychiatric lupus (NPSLE).[2] Whole-transcriptome analysis of the hippocampus in MRL/lpr lupus-prone mice versus wild-type controls revealed transcripts with expression level changes. Subsequent pathway analysis showed enrichment in immune-activated pathways, including microglial phagocytosis and the classical complement pathway, and alterations in genes related to synaptic activity in MRL/lpr mice. Using these results, the researchers investigated the role of the orphan nuclear receptor Nr4a1, which showed reduced expression in MRL/lpr mouse hippocampus. They found that rescuing neuronal Nr4a1 expression protected synaptic engulfment, partially restored abnormal basal synaptic activity, and improved anxiety-like behaviors in MRL/lpr mice. These results illustrate how investigators can use transcriptomics to build molecular profiles and identify targets of interest to unravel the molecular mechanisms behind diseases.
Phenotypic and genotypic association analysis through GWAS
The combination of different sequencing and analysis approaches is more valuable than using independently. A growing application of genome-wide analysis methods is to use them in combination with phenome-wide strategies for the purposes of understanding human disease etiology. A review by Hebbring discusses advances in genome-wide association studies (GWASs) and phenome-wide association studies (PheWASs), and how combined GWAS-PheWAS approaches can reveal new insights about human disease and new drug development opportunities.[3]
GWASs use a “forward genetics” or “phenotype-to-genotype” approach to identify candidate disease/trait associations. They begin with a known phenotype or phenotypes and search large amounts of genomic data for genomic variants (such as single-nucleotide polymorphisms [SNPs]) associated with the known phenotype(s). In contrast, PheWASs use a “reverse genetics” or “genotype-to-phenotype” strategy that begins with a known genetic variant or variants and examines records from varied phenotypes to identify candidate associations across the phenome. PheWAS data is obtained from sources such as electronic health records (EHRs) and biobanks. In this way, PheWAS approaches can identify variants associated with different phenotypes. PheWASs are thus uniquely well-suited to evaluate pleiotropy—to not only identify novel associations and characterize pleiotropic effects, but also evaluate confounding effects within phenotypic information. These abilities ensured PheWAS strategies useful for drug development because pleiotropic variants associated with more than one disease may suggest repurposing opportunities or new indications for existing drugs; may identify additional targets of interest; or may assist in identifying adverse events. GWAS and PheWAS strategies can also be combined in one “GWAS-by-PheWAS” experiment in which a GWAS evaluates large numbers of diseases while a PheWAS evaluates large numbers of variants. Therefore, when thinking about disease research, investigators should consider how sequencing technologies can be used in the context of both genotypic and phenotypic information.
Figure 2 Genome-wide association study in humans
Sequencing methods also have considerably utility for providing vast amounts of genetic information that can be used to compare differences in characteristics among individuals. For example, developing methods for early detection of cancer, when patients are still asymptomatic is an important goal, but demonstrating the effectiveness of such a method requires following a large number of subjects over multiple years, and successfully coordinating genotypic and phenotypic information. Chen et al. developed PanSeer, a noninvasive blood test for cancer-specific methylation signatures from circulating tumor DNA (ctDNA) methylation.[4] The test generates approximately 2 million NGS sequence reads covering a minimum of 200,000 mapped unique DNA molecules to examine 11,787 CpG sites across 595 genomic regions. To evaluate its effectiveness, they used stored plasma samples from the Taizhou Longitudinal Study in which 123,511 healthy patients were followed for cancer occurrence. Chen et al. also developed a machine learning method (classifier) to classify samples as coming from healthy patients or cancer patients. Overall, the classifier sensitivity in post-diagnosis cancer patients was 88%, with 96% specificity, and 95% sensitivity in pre-diagnosis cancer patients. Thus, the PanSeer method successfully detected five cancer types, regardless of tissue of origin, using a common set of methylation markers. This suggests that the classifier genes represent a core epigenetic signature common across multiple cancer types, which warrants further investigation. Moreover, as the authors point out, the assay most likely is not predictive, but rather identifies asymptomatic patients with early-stage cancers who are not identified by current standard methods.
Overall, the use of WGS represents a major breakthrough in the diagnosis, treatment, and prevention of rare and complex diseases. It can be used in context of both genotypic and phenotypic information to uncover the genetic basis. As our understanding of the human genome continues to improve, we can expect WGS to become an increasingly important tool in the fight against rare diseases, and have the potential to revolutionize medicine in the near future.
References
1. Ibañez K et al. Whole genome sequencing for the diagnosis of neurological repeat expansion disorders in the UK: a retrospective diagnostic accuracy and prospective clinical validation study. Lancet Neurol. 2022;21;234-45.
2. Han X et al. Neuronal NR4A1 deficiency drives complement-coordinated synaptic stripping by microglia in a mouse model of lupus. Signal Transduct Target Ther. 2022;7. doi.org/10.1038/s41392-021-00867-y.
3. Hebbring S. Genomic and phenomic research in the 21st century. Trends Genet. 2018;35;29-41.
4. Chen X et al. Non-invasive early detection of cancer four years before conventional diagnosis using a blood test. Nat Commun. 2020;11;3475.