Tools (GO & KEGG) for Gene Set Enrichment Analysis (GSEA)
Gene Set Enrichment Analysis (GSEA) is an important tool in genetic research because it can help researchers identify key biological pathways and processes that are associated with a particular phenotype or disease. GSEA is usually employed in genetic research in the following ways:
- Identifying gene signatures: By analyzing gene expression data using GSEA, researchers can identify gene signatures that are associated with specific phenotypes or diseases. These gene signatures can then be used as diagnostic or prognostic markers, or as potential targets for therapeutic interventions.
- Understanding disease mechanisms: GSEA can be used to identify biological pathways and processes that are dysregulated in a particular disease or phenotype. This information can help researchers understand the underlying mechanisms of the disease, and can lead to the identification of new therapeutic targets.
- Drug discovery: GSEA can be used to identify compounds or drugs that are likely to be effective in treating a particular disease or phenotype. By analyzing the gene expression profiles of cells treated with different drugs, researchers can identify drugs that target specific biological pathways or processes that are dysregulated in the disease.
Firstly, the statistical methods commonly used in enrichment analysis include cumulative hypergeometric distribution, Fisher’s exact test, etc. Since a large number of tests (multiple tests) are usually performed simultaneously in enrichment analysis, the test results need to be corrected using multiple test correction methods to make the results more accurate. These methods include Bonferroni correction to counteract the multiple comparisons problem and Benjamini-Hochberg Procedure for false discovery rate correction. The use of enrichment analysis methods to do bioinformatics research on gene annotation databases has generated many enrichment analysis tools, such as DAVID online analysis tool, R Cluster-Profiler package, Meta-scape, etc. These tools play an important role in facilitating the analysis of gene function and the study of biological knowledge data generated by high-throughput sequencing technologies.
The most common GSEA methods currently used are based on enrichment analysis of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG). Firstly, various techniques are used to multiply a large number of genes of interest, such as differentially expressed gene sets, gene co-expression networks, protein complex gene clusters, etc. In the next step the nodes in GO or pathways of KEGG that are significantly enriched by these gene sets of interest are searched for. This helps in further in-depth and detailed experimental studies. In summary, enrichment analysis is used to decipher the biological knowledge expressed in a set of genes and reveal their roles inside or outside the cell.
Gene Ontology (GO):
The gene ontology (GO) database is a structured standard biological model built by the GO organization in 2000. This model describes our knowledge of biological domains in three aspects that are cellular components, molecular functions and biological processes. It is one of the most widely used gene annotation systems. Each node in the annotation system is a description of a gene or protein. A strict “parent-child” relationship is maintained between the nodes. Thus, a gene or protein can be annotated from three levels.
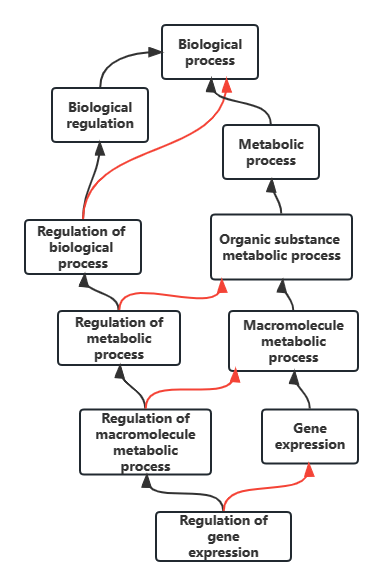
Fig 1: GO flowchart
- MF: Molecular Function
The molecular activities carried out by gene products. This level focuses on the actions performed rather than the entities that are performing the action. - CC: Cellular Component
The location where gene products are active and perform a function. This aspect of GO focuses on cellular anatomy rather than processes. - BP: Biological Process
The larger processes accomplished by the activities of multiple gene products like DNA repair etc. It is to be noted that pathway is not equivalent to biological process. GO does not try to represent the dynamics or dependencies that would be required to fully describe a pathway.
Kyoto Encyclopedia of Genes and Genomes (KEGG)
KEGG is a database for systematic analysis of gene function and genomic information, integrating genomic, biochemical, and phylogenetic information. KEGG is used to understand high-level functions and utilities of the biological system. This database helps researchers study gene and expression of information as a whole. At present, KEGG contains 19 sub-databases. Enrichment analysis is commonly used in KEGG Pathway (It is a collection of manually drawn pathway maps that represent knowledge of the molecular interaction, reaction and relation network). These pathways cover a wide range of biochemical processes.
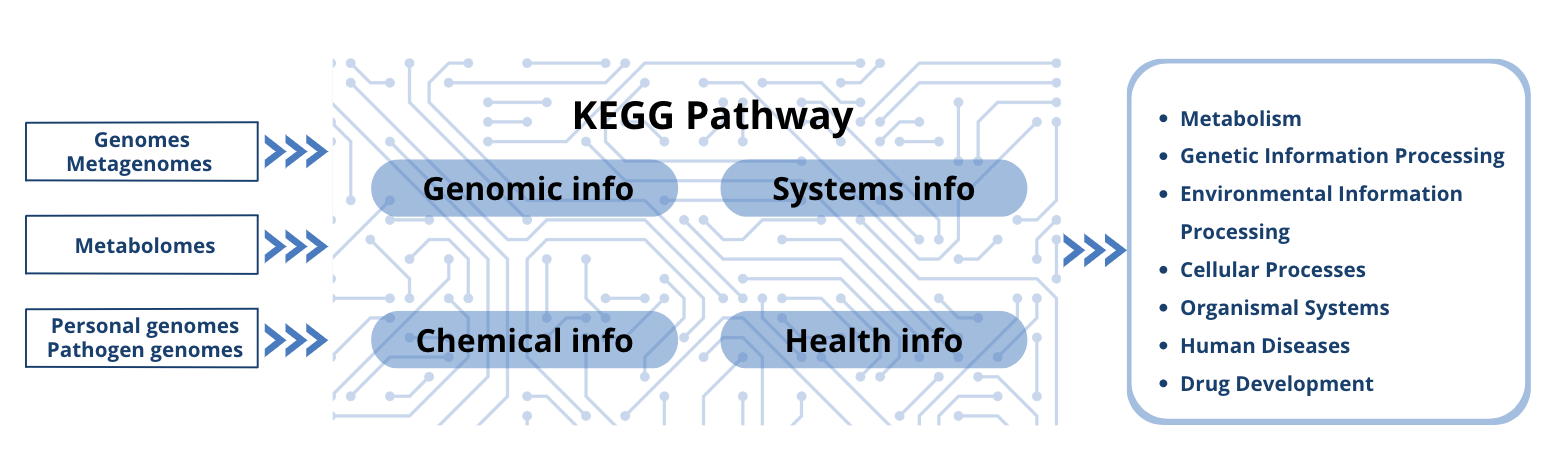
Fig 2: KEGG flowchart
In conclusion, GO and KEGG are the types of GSEA that are the most frequently used for functional analysis. They are typically the first choice because of their long-standing curation and availability for a wide range of species. They can all be processed through Novomagic’s online tools with just a click.