RNA-Seq Results Explained: What can you expect from the analysis?
RNA-seq is a genomic tool that uses next-generation sequencing (NGS) technologies to examine the presence and quality of RNA in a biological sample. This technique allows us to study the transcriptome of a cell at a given time and has a wide variety of applications, from examining gene expression to genome annotation. As a result, this tool can be used to answer a range of biological questions, such as examining how a cell responds to a specific environment or looking at pathogen-host interactions. In the following article, we will build on the information provided in our webinar.
RNA-seq Analysis
RNA-seq can produce a massive amount of data, with millions of reads that need to be quantified, mapped, and interpreted before we can begin to understand what the data from the transcriptome is showing. At Novogene, there are five main steps that we take to analyze this data:
- Quality Control
- Mapping
- Quantitative Analysis
- Functional Analysis
- Qualitative Analysis
First, the sequencing data needs to go through quality control. The raw data is filtered to remove low-quality reads as well as sequences that contain adapters. This step enables us to provide a clean, high-quality data set that can then be mapped to the reference genome.
Three types of reads are obtained from the RNA-seq data: (1) sequences that map entirely within an exon, (2) reads that span over 2 exons, and (3) reads that span over more than 2 exons. To visualize this, we use a software called HISAT2. This uses a graph-based approach to index the reference genome. The resulting BAM file can be viewed using the Integrative Genomics Viewer (IGV), which shows you the position of the reads within the genome, as well as the read abundance, demonstrating which genes are being differentially expressed within your sample.
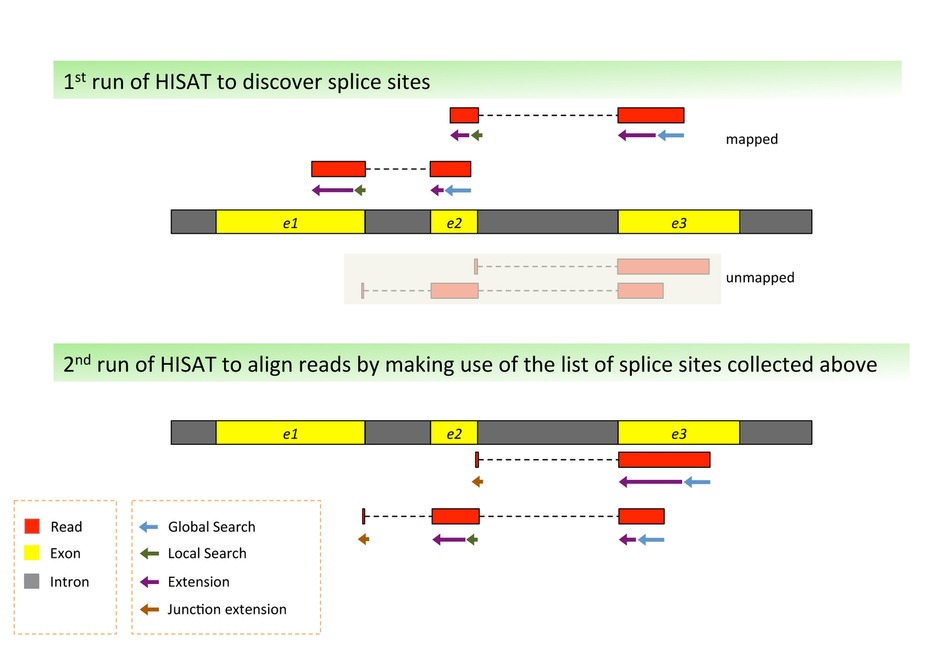
Figure 1 the Algorithm of Split Reads Comparison by HISAT2
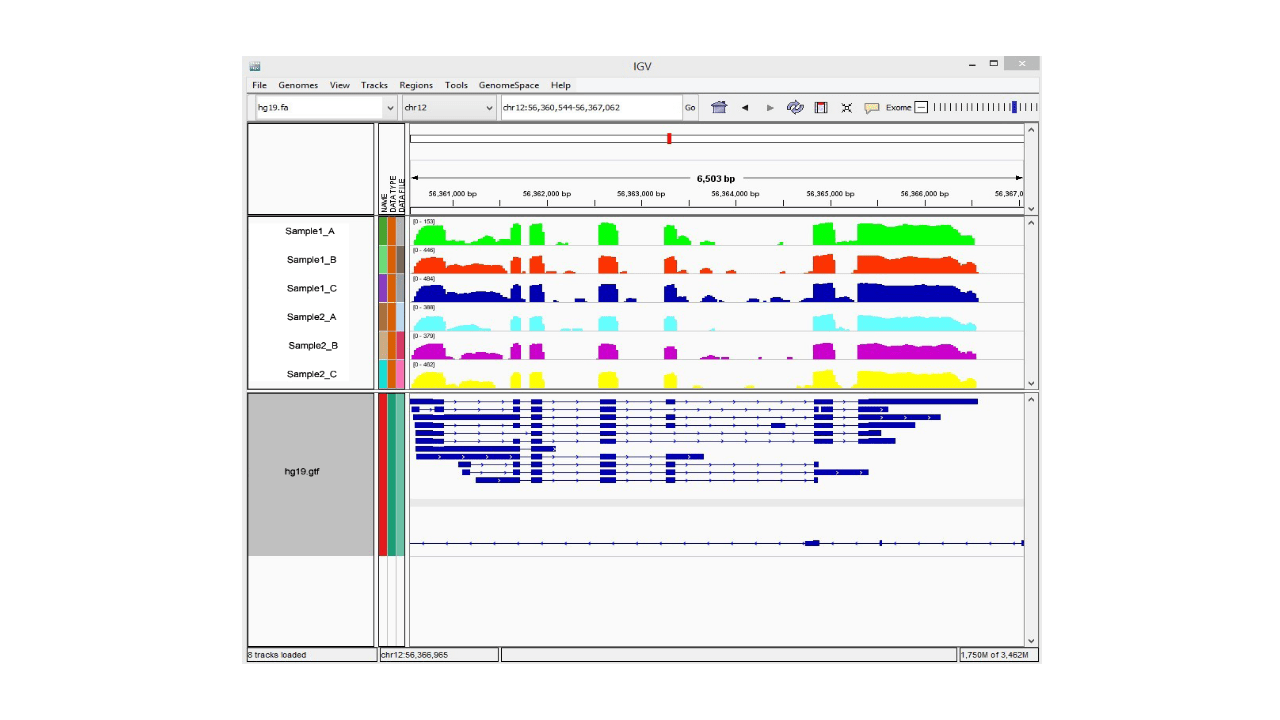
Figure 2 IGV Browser Visualization Result
The next step is to count how many reads align to each gene, and we do this using a program called Feature Counts. This program summarizes the number of reads that map to a single location and from here, the reads are then assigned to a gene. The RNA-seq data must then be normalized to account for the total reads sequenced for each sample or the read depth. A Pearson’s correlation or Principle Component Analysis can then be carried out on this normalized data to check for intro- and intergroup sample variability and any outliers.

Figure 3 Principal Component Analysis result
To check for differentially expressed genes, we use a software called DESeq2. Briefly, this software uses the raw data to calculate a group mean and a global mean. It then calculates the deviations of the group mean from the global mean to determine if any changes in gene expression are significant. The output from this provides you with p-values to determine the significance as well as the gene name, location, and description of its function. Different diagrams can then be used to visualize this information, such as a volcano plot or a cluster analysis. These visual tools use different colors to demonstrate which genes have been down-regulated as well as those that show a significant increase in their expression, providing a simple and easy to interpret visualization of your RNA-seq data.
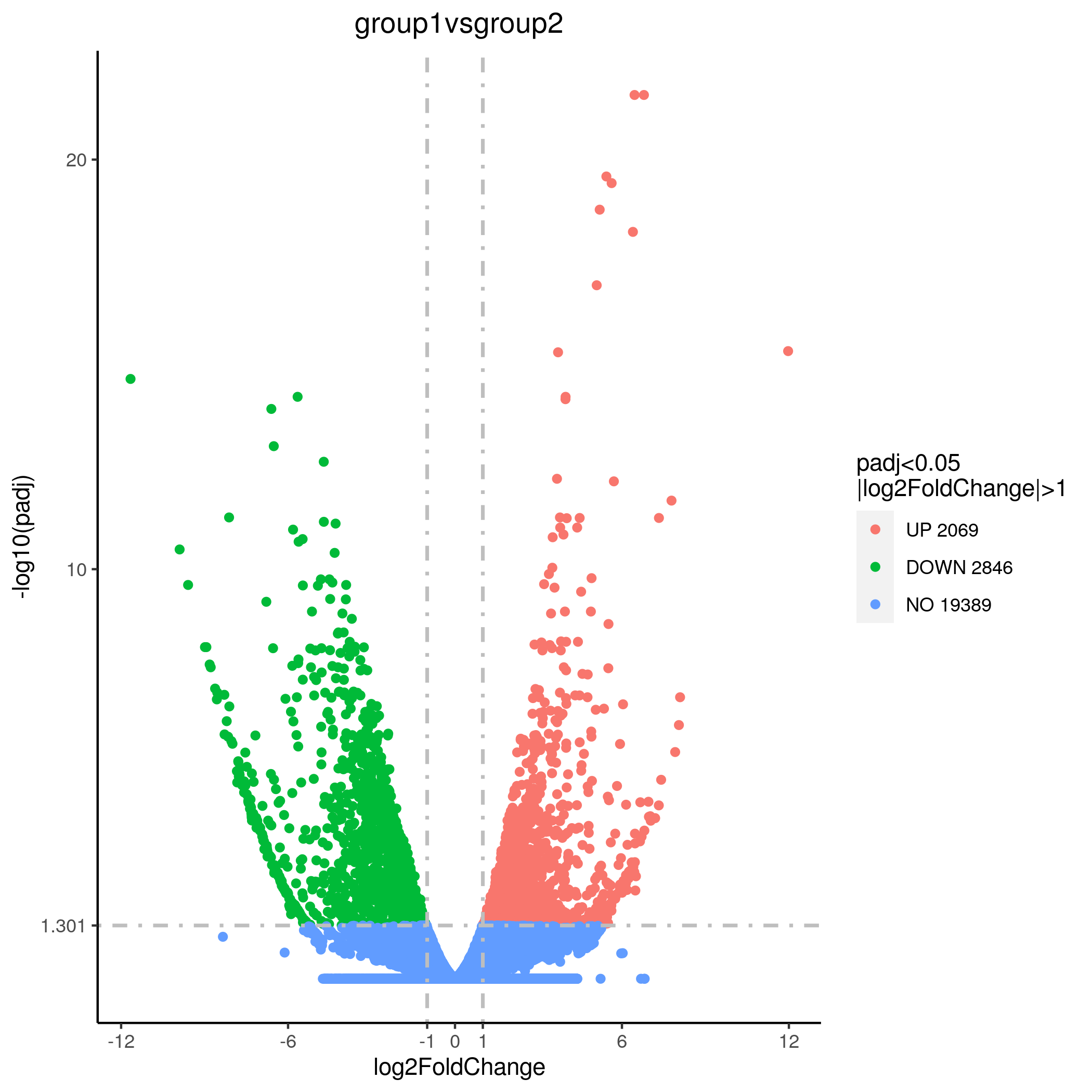
Figure 4 Volcano Map of Differentially Expressed Genes
Once we have this information available, we can then assign a biological function to the genes that have been differentially expressed using a program called Cluster Profiler. The information provided in this step could be used to infer what pathways or functions the genes are involved in or identify trends in gene expression, which could be used to identify novel pathways. Finally, we move on to the qualitative analysis step, which can be used to characterize the transcripts. Here we can use several programs to carry out further analysis, such as variant discovery, alternative splicing, and fusion transcript analysis.
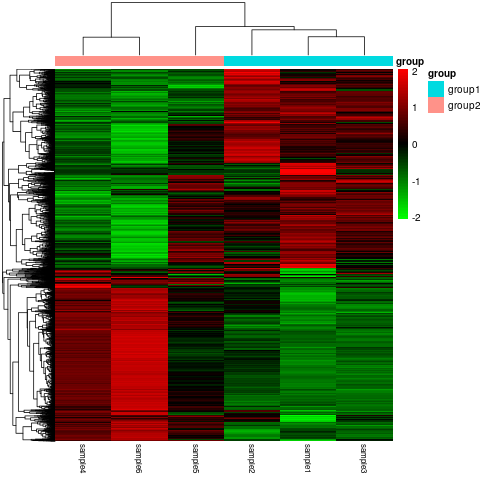
Figure 5 Differential Expression Gene Clustering Heatmap
Why choose RNA-seq?
RNA-seq is a powerful tool used to quantify the transcriptome profile of a cell at any given time. To learn more about what you can expect from RNA-seq analysis as well as the services that Novogene provide, listen to our webinar available here: https://www.novogene.com/us-en/resources/webinar/rna-seq-results-explained-what-you-can-expect-from-the-analysis/
Why Novogene?
At Novogene, we use the latest technology to provide you with high-quality data in a short amount of time. In addition, our bioinformatics experts are on hand to help you with all your analysis requirements, from examining gene expression to SNP and InDel analysis, functional enrichment analysis, fusion gene prediction, alternative splicing analysis, and more. Our comprehensive services can provide you with high-quality results and publication-ready figures and tables.
In addition to the services described above, we also offer other RNA-seq solutions. These include de novo transcriptome assembly for organisms where a reference genome is not available and Pac-Bio Iso-seq, which uses long reads to provide a more complex analysis. This is particularly useful for de novo transcriptome assembly.