Next generation sequencing and its clinical applications
Next-generation sequencing (NGS) is the current generation of sequencing, which is more time and cost-efficient than traditional sequencing technologies. The NGS has advanced and changed our perspective on investigating genomics and its applications. From the human genome project in 2001 to chip-seq in 2007, the sequencing revolution for cancer research in 2008, the cataloging public genome encyclopedia in 2012, and current days’ short and long read latest sequencing are some of the milestones have been achieved in genomics research.
Evolution of DNA sequencing tools
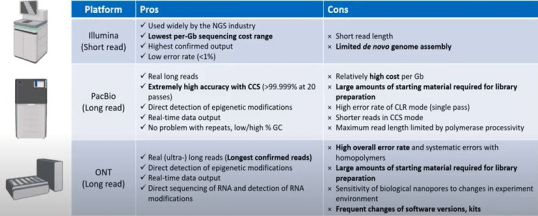
Short and long read sequencing are divided into three generations: first, second and third. Sanger sequencing (1st generation), which can sequence 500-1000bp fragments, involves selected incorporation of chain-terminating dideoxy nucleotides by DNA polymerase during in vitro DNA replication. The modified dNTPs are then visualized on an electrophoresis gel. Illumina (2nd generation), on the other hand, sequences 50-500bp. It massively parallels acoustic technology that offers ultrahigh throughput statement scalability and speed. Both Sanger and Illumina are considered as shortly sequenced tools. Recently, a third-generation sequencing called Nanopore sequencing was developed to sequence long reads (tens of KB fragments). It is the only sequencing technology that offers real-time analysis. Different sequencing platforms can be compared, as shown in the following diagram.
Multiomics Approaches
In Genomics, three different types of sequencing are used, namely, Whole genomic sequencing (WGS), Whole exome sequencing (WES), and Target region sequencing (TRS). WGS includes two different approaches: De novo sequencing and Re-sequencing. No reference sequence genome exists in the former, while a reference genome already exists in the latter. On the other hand, WES focuses on the coding regions of the genome. It sequences all the protein-coding regions of the genome known as the exome. By sequencing only 2%, it can identify 85% of the known disease-related variants. It is cost-effective for WGS and quickly detects rare and low-frequency mutations. The third one, TRS, focuses on essential target genes. It requires less sample input and detects known and novel variants within your region of interest. Hybridization capture and amplicon sequencing are two popular methods used in this sequencing.
Epigenomics involves understanding genetic control beyond DNA sequence. RNA- sequencing has practical applications parallel to DNA sequencing. Transcriptomics is the study of RNA transcripts produced by the genome. RNA-seq enables the sequencing of aberrant RNA by illuminating the functional consequences of uncertain sequence variants. This leads to a better understanding of the cause-and-effect relationship of variation and helps us prioritize variations for diagnosis and treatment. Clinical epigenetics is used to treat patients with cancer, neurological diseases, infectious diseases, and problems with the immune system.
Clinical applications
Genomic applications can significantly impact clinical care in many ways, from diagnosing problems before birth to caring for people at the end of their lives.
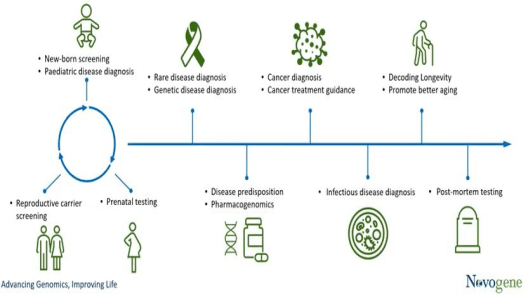
Disease genomics generally deals with severe monogenetic diseases (typically 1-2 variants per family) and common complex diseases (massively polygenic). Disease-causing variants also form a spectrum of frequency and effect, such as rare, low-frequency, and common variants. There are about 7000 rare diseases, with more discovered every year. An estimated 300 million people suffer from a rare disease. Some examples are Tay-Sachs disease, Gaucher disease, and Huntington’s disease. 80% of rare diseases are genetic or have a genetic component.
Most rare diseases result from mutations in a single gene, transmitted according to a Mendelian pattern of inheritance, hence are known as ‘Mendelian’ diseases. Following WGS and WES, many variants are classified as candidates for a disease, and pedigree analysis can help prioritize such variants. After pedigree analysis, linkage analysis is another approach to increase the ability to identify causal variants. It is the co-segregation of a chromosomal region and a trait of interest. It uses family data to find genetic loci that could have genes that make people more likely to get sick.
On the other hand, the most critical changes in healthcare right now are related to common non-communicable diseases like autoimmunity, neurodegeneration, and heart diseases. These disorders are caused by how a person’s genes and environment work together. There are hundreds of common genetic variations, each with negligible effect on the phenotype. Together, these variations add to the complexity of common diseases, which are also complex.
Genome-wide association studies (GWAS) look for changes linked to disease by comparing the genotypic or allelic frequencies of people with and without the disease. Some complicated conditions for which effective treatments have been found include type 2 diabetes, Parkinson’s disease, several types of heart disease, and many types of cancer. Another application is low pass whole genome sequencing, known as low-coverage WGS (lcWGS) or shallow WGS (sWGS). It produces a mere fraction of the data and relies on computation methods.
Moreover, cancer genomics uses sequencing technology to identify the DNA and RNA alterations by comparing the sequencing of cancerous cells to normal body tissues. Understanding different mutations, such as germline, somatic, driver, and passenger mutations, is necessary for cancer genomics. Emerging clinical applications of cancer genomics include monitoring treatment responses and characterizing resistance mechanisms.
While investigating cancer, tumor heterogeneity is an important concept that needs to be considered. It means that different tumor cells show different morphological and phenotypic profiles. So, intertumor or intratumor heterogeneity can be observed, affecting cancer pathways, driving phenotypic variations, and significantly changing personalized cancer medicine.
Also, Tumor mutational burden (TMB) is used as a potential biomarker in cancer treatment. It is associated with immune checkpoint inhibitor (ICI) therapy. ICIs can stimulate and allow the immune system to detect the neo antigens (formed due to changes in DNA and proteins) and destroy the tumor.
Testing approaches also identify damage and therapeutic options for speeding early detection. So, two main biopsy approaches are used to take somatic mutations cancer: tumor biopsy and liquid biopsy. The former looks at mutations in the DNA of cancer cells, whereas the latter looks at small pieces that are shed by cancer cells into the bloodstream. A new method, germline testing, uses blood or saliva to get DNA for sequencing.
Genomics helps in unlocking personalized medicine. Genomics-powered precision health can improve outcomes, help pinpoint the underlying causes, and optimize treatments.
There are other relatable applications, such as Human leukocyte Antigen (HLA) typing. HLA genotyping is individuals’ HLA class 1 and class 2 gene polymorphisms. HLA system encodes the major histocompatibility proteins in humans, which regulate the immune system.And the final one is mitochondrial genomics. Mitochondrial disorders are heterogenous diseases that can arise at any stage of life. So, Mitochondria genomics is used to study human diseases and population genetics and is essential for identification and forensics applications.
How Novogene can help
Novogene’s mission is to advance genetics and improve life. Our services apply to a wide range of real-world situations, such as cancer, forensics, food and water safety, biopharma, and industrial diagnostics. With reduced human error and delay, Novogene’s cutting-edge lab, the latest Novaseq 600 sequencing technology, Falcon II sequencing delivery system, and supercomputing capacity provide faster results. We offer human, microbial, plant, and animal genome sequencing, whole genome and transcriptome analysis, and epigenome analysis.