Metagenomics: Host DNA Removal or Not?
Over the past decade, a dramatic reduction in DNA sequencing costs has propelled shotgun metagenomic sequencing into the forefront. Shotgun metagenomic sequencing is an indispensable tool for characterizing bacterial populations across diverse environments and host systems, bypassing the need for target-specific primers.
Shotgun metagenomic sequencing uses next-generation sequencing (NGS) technology to provide not only information on the taxonomic annotations of each organism, but also the functional profiling, gene prediction and microbial interaction of the whole community. However, in the analysis of host-derived samples, this broad-spectrum approach poses a challenge. Samples of metagenomic sequencing with higher host content typically include those obtained from host-associated environments or tissue samples where the microbial load is relatively low compared to the host DNA [1-3]. Here are some examples:
- Human or Animal Tissue Samples: Samples collected from organs, body fluids, or mucosal surfaces of humans or animals. Tissue samples obtained from organs such as liver, muscle, heart, and brain often have high levels of host DNA compared to microbial DNA.
- Clinical Specimens: Clinical samples from patients, including blood, urine, and tissue biopsies. These samples may have a higher proportion of host DNA, especially in cases where the microbial load is low, such as in blood infections or sterile body sites.
- Host-Associated Microbiomes: Samples from the human gut, oral cavity, skin, respiratory tract, and reproductive tract. While these sites harbor diverse microbial communities, the host DNA content can be significant, particularly in mucosal samples where host cells are abundant.
- Host-Dominated Environments: Environments where host organisms are the primary source of DNA, such as animal husbandry facilities, animal shelters, and zoos. Samples from these environments may contain a higher proportion of host DNA compared to environmental samples with a more balanced microbial community.
- Host-Pathogen Interactions: Samples from infection sites where host-pathogen interactions occur, such as wounds, abscesses, or infected tissues. The presence of pathogens alongside host cells can further increase the host DNA content in these samples.
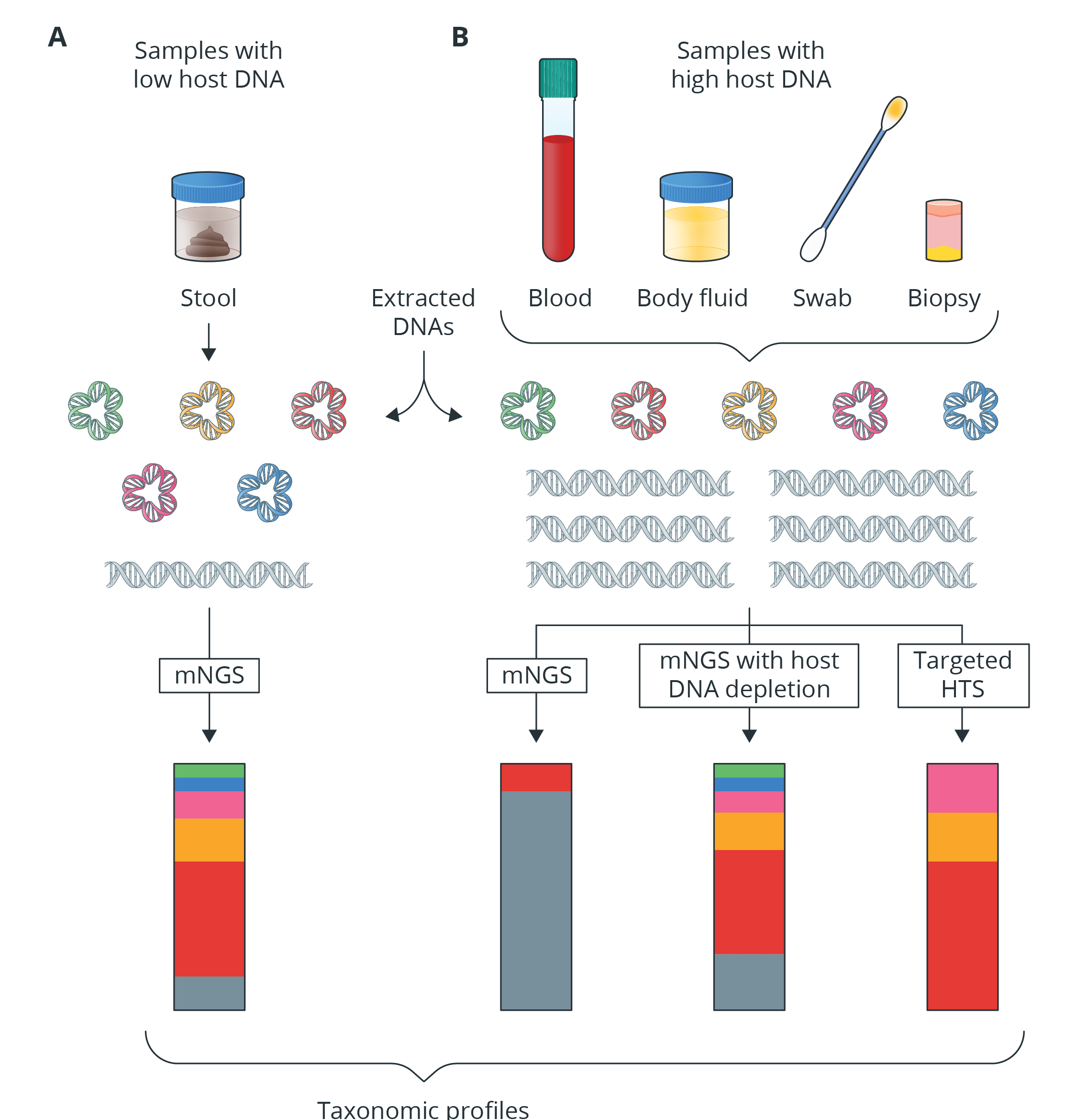
Fig 1. Adapted from Shi et al., 2022c [4]. Removal of host DNA before sequencing can improve the resolution of microbial DNA. Gray represents host DNA; red, yellow and pink represent various bacterial species; and green and blue represent viruses and archaea, respectively. mNGS, metagenomic next-generation sequencing. HTS, high throughput sequencing.
Current Host DNA Depletion Approaches
The development of a reliable method to systematically deplete host reads in shotgun sequencing is imperative for virtually all studies focusing on host-derived microbiomes. Here, we summarized the frequently used host DNA depletion methods [4,5].
- Physical separation (microfiltration and centrifugation): Host cells are significantly larger than microbial cells, making it possible to separate human-derived cells using methods such as membrane filtration, low-speed centrifugation, or flow cytometry (Fig. 2B).
- Microbial DNA enrichment: This methodology leverages the discrepancy in cytosine methylation frequency between eukaryotic and prokaryotic DNA. Utilizing MBD-Fc-bound magnetic beads, CpG-methylated host DNA sequences can be selectively captured and removed by a magnetic field while leaving the non-CpG-methylated microbial DNA in the supernatant available for downstream applications (Fig. 2C).
- Enzymatic and chemical treatments (selective host cell lysis and DNase/PMA treatment): The prevailing pre-extraction method for eliminating host DNA involves initially subjecting human cells to a selective lysis buffer (i.e., saponin), followed by treatment with DNA nucleases (Fig. 2D) or propidium monoazide (PMA) as photoreactive DNA-binding dye (Fig. 2E).
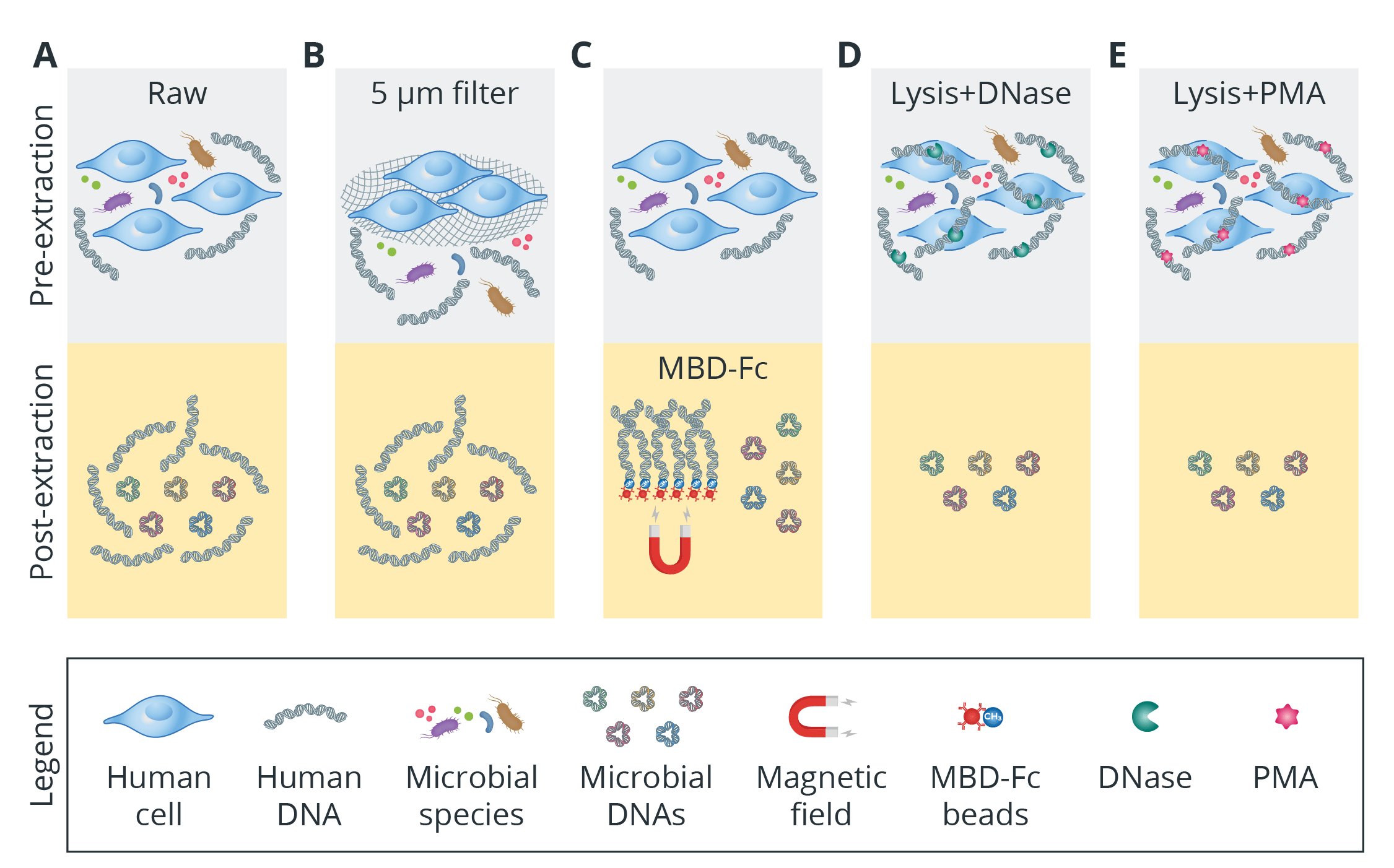
Fig 2. The efficiency of five methods of host DNA depletion for shotgun metagenomic sequencing. MBD stands for methyl-CpG binding domain of human MBD2 protein that is fused to the Fc fragment of human IgG antibody. Adapted from [5].
- Bioinformatic approaches:
- Sequence alignment for removal: This method involves aligning metagenomic sequencing data with known host genome sequences and removing sequences that are highly similar to the host genome. This requires efficient alignment software such as Bowtie2 [7], BWA [8], etc.
- K-mer based methods: Independent of reference genomes, this approach identifies and removes host sequences by comparing the frequency distribution of short sequence fragments (k-mers). This method is suitable for situations where the host genome is unknown or incomplete [6, 9].
In summary, a combination of various methods and technologies is used to increase the efficiency of host DNA removal [4]. For example, physical separation might be used initially to reduce the bulk of host cells, followed by enzymatic treatment to degrade any remaining host DNA. Finally, bioinformatic subtraction can clean up any residual host sequences that make it through to sequencing. Each method has its context of application, depending on the type of sample, the relative abundance of host vs. microbial DNA, and the specific goals of the study. The choice of method can significantly impact the cost, complexity, and sensitivity of metagenomic analyses.
Novogene’s Metagenomics Host DNA Removal Services
Novogene adeptly eliminates host DNA through a two-step process: initially, host cells undergo selective differential lysis, followed by the enzymatic digestion of the liberated host DNA. The host cells can be selectively lysed by adjusting the pH and temperature of the lysing solution, without damaging microbial cells. And the efficacy depends on the differential susceptibility of host and microbial cells to the lysis conditions. Once the host DNA is released, it can be degraded by DNase, or inactivated by covalently bound PMA (Fig. 3).
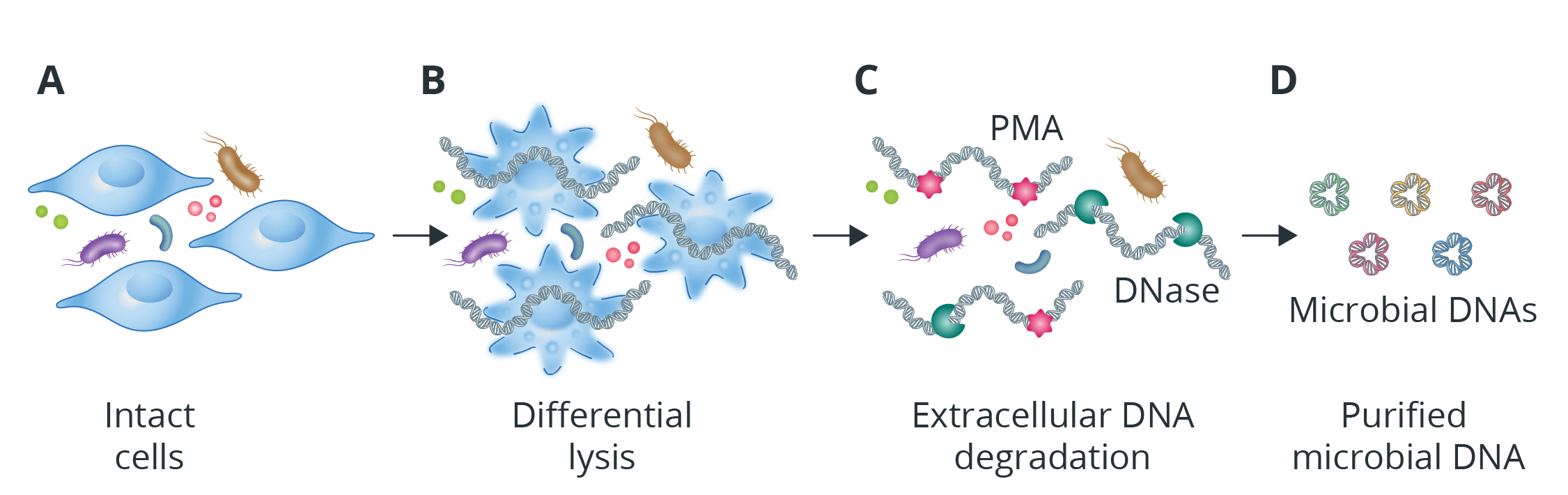
Fig 3. Novogene’s metagenomic host DNA removal services before sequencing.
A synergistic approach combining mechanical and chemical lysis ensures the effective disruption of intact cells, facilitating the purification of microbial DNA. To address any remaining host sequences that persist into the sequencing phase, bioinformatic subtraction is employed as a precise cleanup strategy.
Key Advantages of Novogene’s Shotgun Metagenomic Sequencing
- Outstanding end-to end service with fast-turnaround time, top-notch quality and cost-effective pricing.
- Expert bioinformatics analyses provide comprehensive data on annotated genes, metabolic pathways and antibiotic resistance genes profiles.
- Our strategy synergizes both deep shotgun sequencing and efficient shallow shotgun sequencing on short-read platforms, serving diverse applications to unveil comprehensive analysis.
Reference
[1] Human Microbiome Project Consortium. “A framework for human microbiome research.” Nature vol. 486,7402 215-21. 13 Jun. 2012, doi:10.1038/nature11209
[2] Chiu, Charles Y, and Steven A Miller. “Clinical metagenomics.” Nature reviews. Genetics vol. 20,6 (2019): 341-355. doi:10.1038/s41576-019-0113-7
[3] Heravi, Fatemah Sadeghpour et al. “Host DNA depletion efficiency of microbiome DNA enrichment methods in infected tissue samples.” Journal of microbiological methods vol. 170 (2020): 105856. doi:10.1016/j.mimet.2020.105856
[4] Shi Y, Wang G, Lau HC, Yu J. Metagenomic Sequencing for Microbial DNA in Human Samples: Emerging Technological Advances. Int J Mol Sci. 2022;23(4):2181. Published 2022 Feb 16. doi:10.3390/ijms23042181
[5] Marotz, Clarisse A et al. “Improving saliva shotgun metagenomics by chemical host DNA depletion.” Microbiome vol. 6,1 42. 27 Feb. 2018, doi:10.1186/s40168-018-0426-3
[6] Langmead, Ben et al. “Ultrafast and memory-efficient alignment of short DNA sequences to the human genome.” Genome biology vol. 10,3 (2009): R25. doi:10.1186/gb-2009-10-3-r25
[7] Li, Heng, and Richard Durbin. “Fast and accurate short read alignment with Burrows-Wheeler transform.” Bioinformatics (Oxford, England) vol. 25,14 (2009): 1754-60. doi:10.1093/bioinformatics/btp324
[9] Wood, Derrick E, and Steven L Salzberg. “Kraken: ultrafast metagenomic sequence classification using exact alignments.” Genome biology vol. 15,3 R46. 3 Mar. 2014, doi:10.1186/gb-2014-15-3-r46