GEO data mining (IB) – Downloading Sequence Read Archive raw data
Life science researchers may be interested in performing bioinformatics research or in incorporating bioinformatics analysis into their research programs, but they may not be sure which gene is the best starting point. Alternatively, they may have a gene of interest, but may be unsure of how to find disease research data that can be used for further analysis.
The Sequence Read Archive (SRA) database is an excellent source of raw sequencing data that can be mined to address those questions or uncertainties. For example, SRA data can be a source of potentially interesting genes from high-scoring publications with source data that can be downloaded for in-depth mining analysis, and then bioinformatics results can be verified with “wet lab” experiments.
In Part IA , we covered how to download Gene Expression Omnibus (GEO) data. However, using GEO expression matrix data to analyze up- or down-regulation of genes is not the only source of innovative research, and some “star” genes in the GEO database may have already been thoroughly analyzed. Original published raw sequencing data in the SRA database is another source of great potential waiting to be tapped. Investigators who are used to traditional work on coding genes can use SRA data to enrich their studies with non-coding genes and explore DNA regulatory elements. If the sequencing depth is very deep, trans-shearing can be studied to mine potential circular RNAs. Even the original sequencing data can be analyzed from scratch to explore new genes. Therefore, combining the GEO database and SRA database for data mining enables investigators to explore gene functions and pathways expansively and in greater detail than if they used only one of the two databases.
Introduction of SRA Database
The SRA database is an NCBI (National Center for Biotechnology Information) sub-library for storing high-throughput sequencing data. The SRA database collects original sequencing data, and the original sequencing data from published articles around the world can be downloaded for free. The basic framework of the SRA database uses four types of metadata: STUDY, SAMPLE, EXPERIMENT, and RUN.
- STUDY: corresponds to a research topic or research project. These have the prefix “SRP,” “DRP,” or “ERP.”
- SAMPLE: one or more samples make up an experiment. These have the prefix “SRS,” “DRS,” or “ERS.”
- EXPERIMENT: includes one or more samples for one or more RUN sequencing results. These have the prefix “SRX,” “DRX,” or “ERX.”
- RUN: corresponds to results. These have the prefix “SRR,” “DRR,” or “ERR”.The rest of this article will focus on downloading RUN files because they contain actual sequence data in FASTQ format.
Downloading SRA library raw data
Raw sequence data can be downloaded from the SRA database in both non-LINUX and LINUX operating environments.
Web-based data downloads for non-LINUX operating environments
1. Web page download
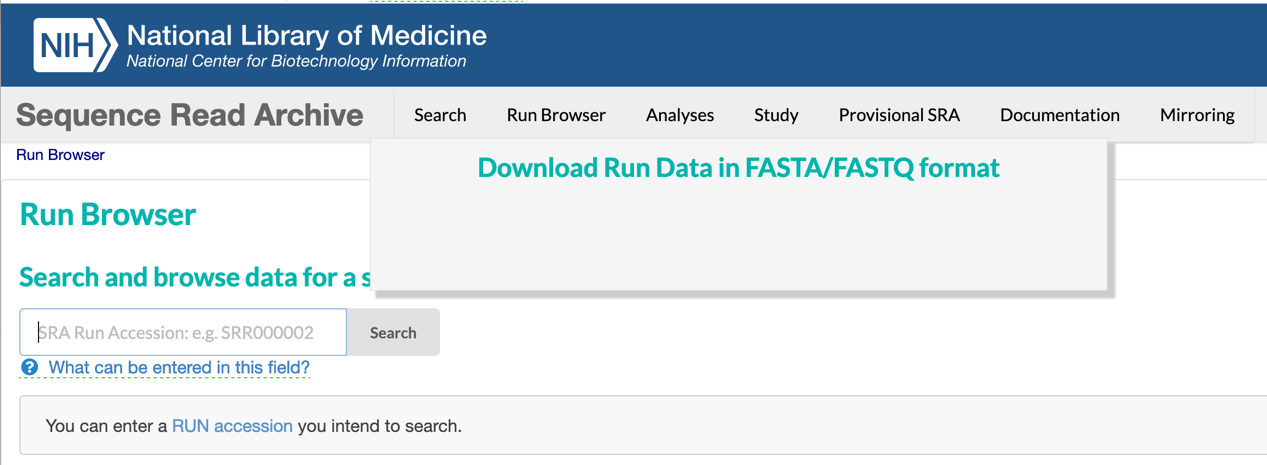
SRA data can be downloaded using any browser. Enter an SRA Run Accession Number into the Run Browser on the SRA website which is currently accessible at: https://trace.ncbi.nlm.nih.gov/Traces/index.html?view=run_browser&display=download.
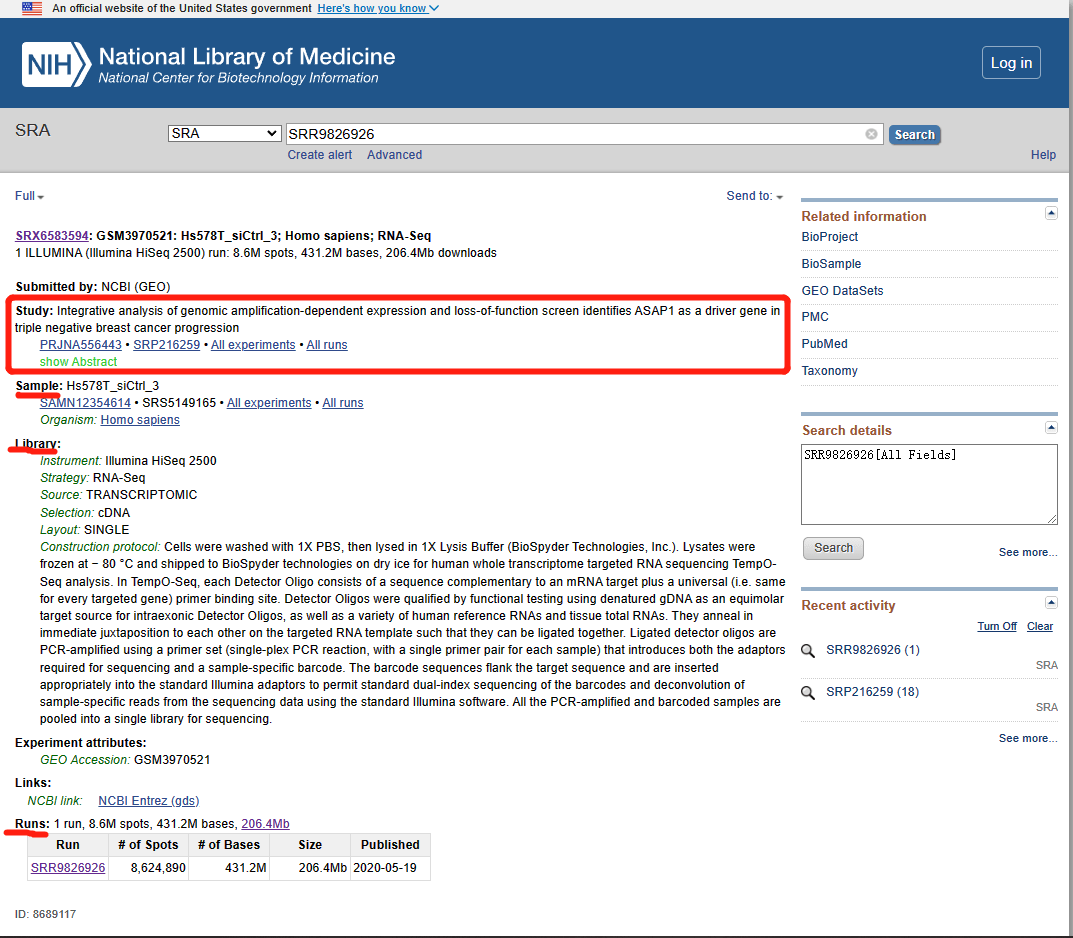
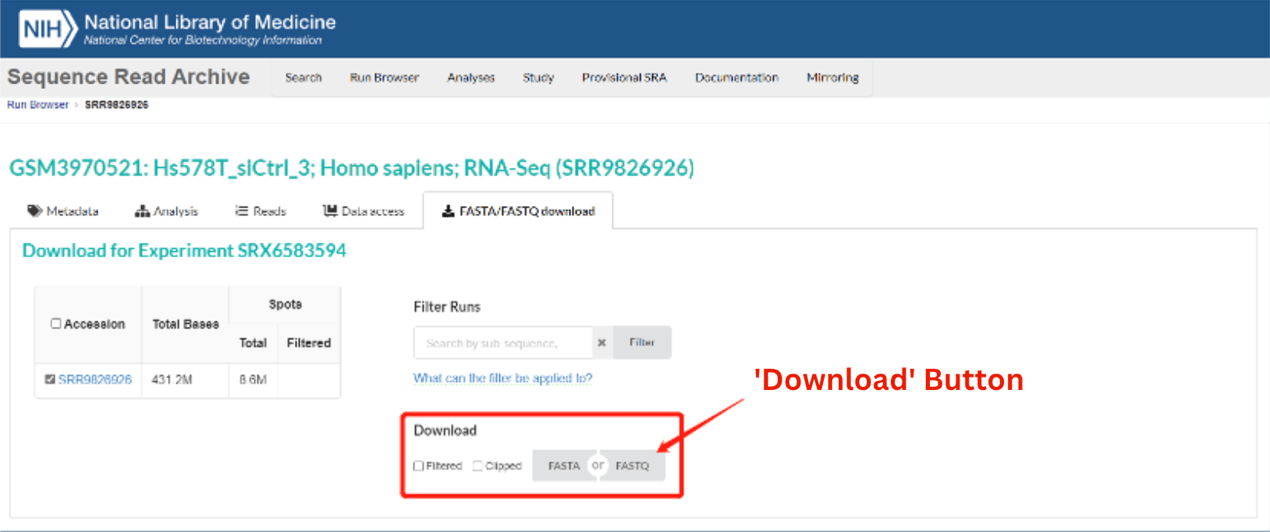
For example, entering SRA Run Accession Number SRR9826926 brings up the record for Experiment SRX6583594
(https://trace.ncbi.nlm.nih.gov/Traces/index.html?view=run_browser&acc=SRR9826926&display=download), as shown in the image below. Sequence data can be downloaded automatically by clicking on either the FASTA or FASTQ buttons
The first letter of a prefix indicates the source database to which the sample was originally uploaded: S, SRA; D, DNA Data Bank of Japan (DDBJ); and E, European Bioinformatics Institute (EBI). The SRA database synchronizes the sequencing data from EBI and DDBJ.
(circled in red).
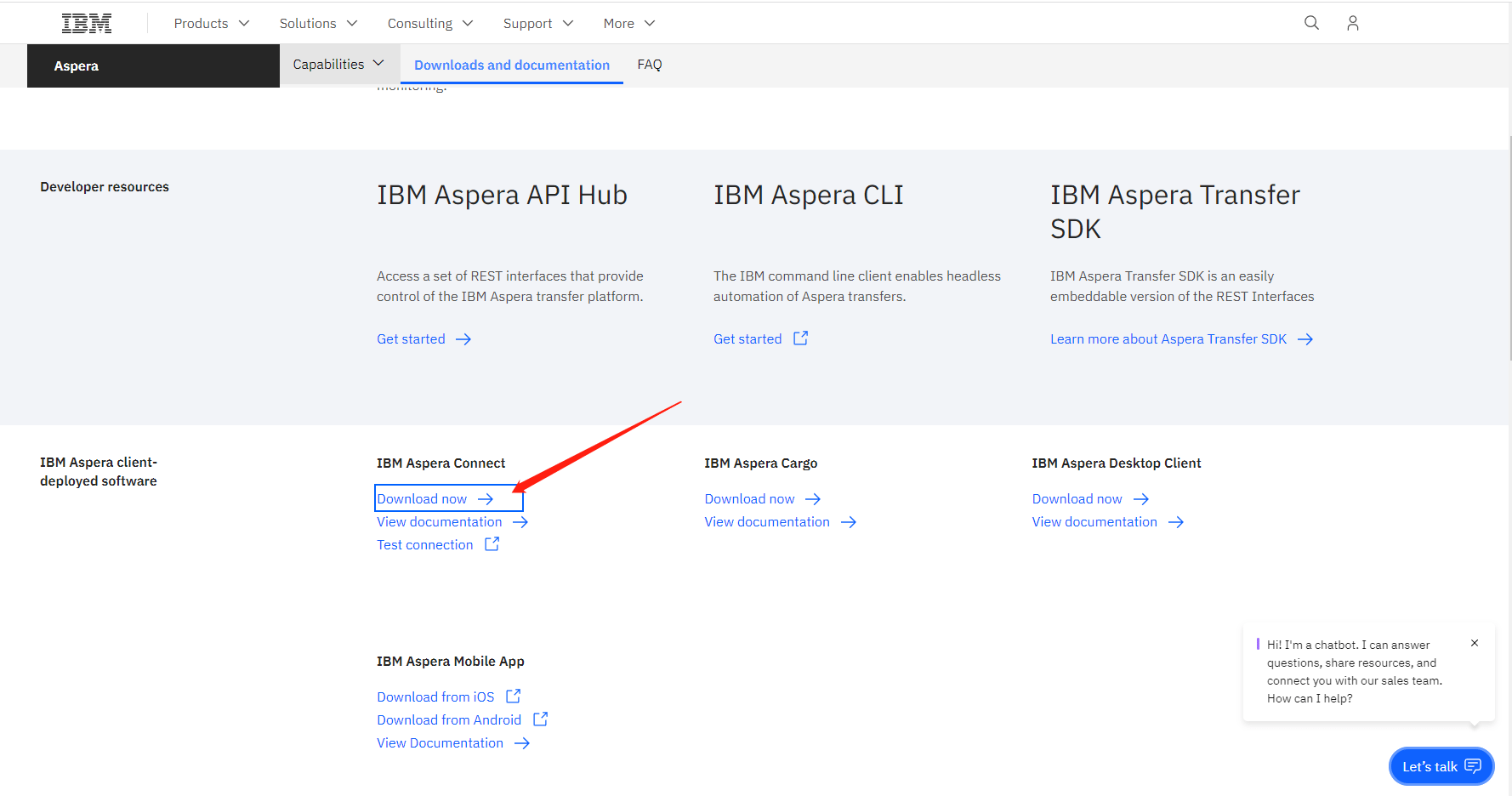
2. Browser plug-ins
The IBM Aspera Connect browser plug-in enables quick downloading of batched SRA data. Download the Aspera connect plug-in (https://www.ibm.com/aspera/connect/) as shown in the image below, the follow the webpage download steps described above.
Downloading SRA data in a LINUX operating environment
SRA data can be downloaded in a LINUX environment using the prefetch command to grab the Accession List information downloaded by SRA (shown in the image below) and then using Aspera Connect to download to the required FASTQ file(s).
Prefetch command download
First, integrate the Accession List to be downloaded through the RUN selector of SRA, and then download SRA files in batches through the Linux command line.
https://www.ncbi.nlm.nih.gov/sra/?term=SRP216259
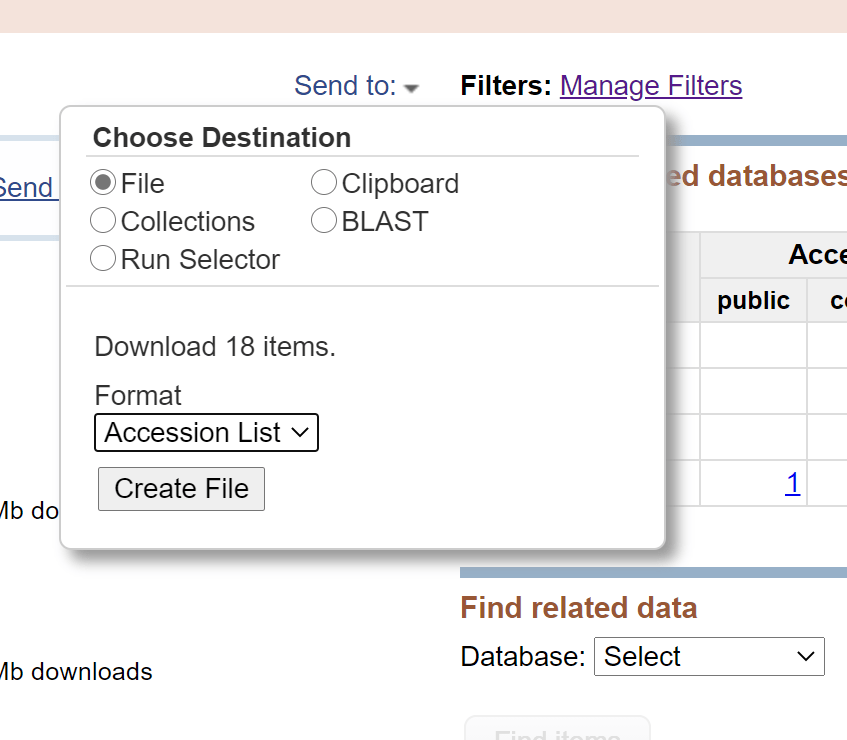
Click the button of ‘Create file’ to get the Accession list
# Use the prefetch command to download a single file: such as SRR1039510
prefetch SRR1039510
# Batch download: create a loop and check
outputdir=/**/sra
cat sampleId.txt | while read id
do
echo “prefetch ${id} -O ${outputdir} ”
done >download.sh
nohup sh download.sh >download.log &
# Verify data integrity
Vdb – validate SRR1039510
Aspera Connect download
Search the European Nucleotide Archive (ENA;
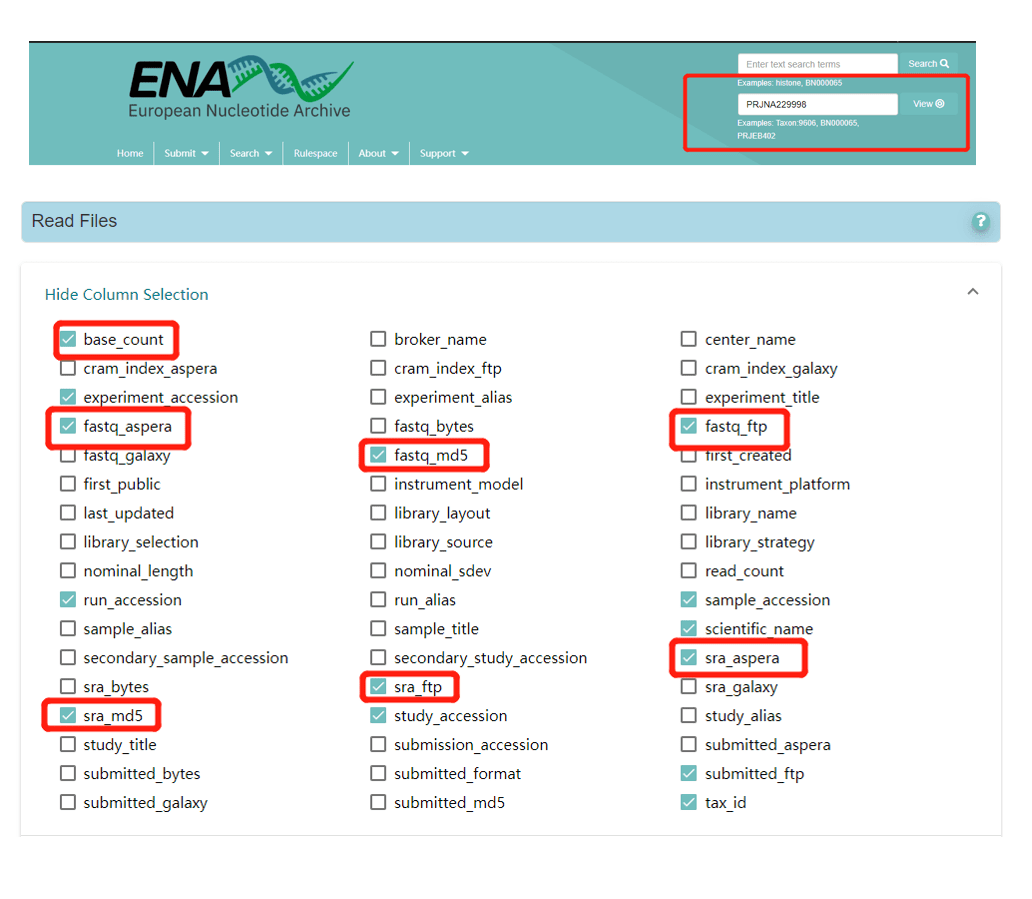
https://www.ebi.ac.uk/ena/browser/home) for a project number of interest to obtain the download address of the relevant FASTQ file(s), and check the required information under Column Selection. For example, the image below shows the Column Selection options for project PRJNA229998 (https://www.ebi.ac.uk/ena/browser/view/PRJNA229998).
Use the following code to download the FASTQ files via Aspera Connect:
# download a single file
# sra format
ascp -k 1 -QT -l 300m -P33001 -i ~/**/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/srr/SRR103/008/SRR1039508 .
# gz format
ascp -k 1 -QT -l 300m -P33001 -i ~/**/asperaweb_id_dsa.openssh era-fasp@fasp.sra.ebi.ac.uk:/vol1/fastq/SRR103/000/SRR1039510/SRR1039510_1.fastq. gz .
# Batch download
# Get the sra.url file, if there are special characters at the end of the line, run sed -i “s/\s*$//g” sra.url to remove the special characters at the end of the line
cat filereport_read_run_PRJNA229998_tsv.txt |awk ‘NR>1{print $NF}’ >sra.url
cat filereport_read_run_PRJNA310728_tsv.xls |awk -F ‘\t’ ‘NR>1 {print $20}’ |tr ‘;’ ‘\n’ >fastq.url
# Order
outputdir=/**/sra
cat sra.url | while read id
do
echo “ascp -k 1 -QT -l 300m -P33001 -i ~/**/asperaweb_id_dsa.openssh era-fasp@${id} ${outputdir}”
done >sra.download.sh
# Submit background
nohup sh sra.download.sh >sra.download.log &
## Data integrity check
# get md5 value
awk ‘NR>1{print $11″\t”$4}’ filereport_read_run_PRJNA229998_tsv.txt >md5.txt
# md5 value test
md5sum -c md5.txt
We have now obtained the raw materials for analysis by downloading sequencing data from the GEO and SRA databases. Stay tuned for the next tutorials in our “GEO Data Mining” series. We will cover analysis of differential gene expression and visualization, pathway enrichment analysis, and more.