Introduction to Microbial Whole Genome Sequencing
Microbial Whole Genome Sequencing is a critical approach for sequencing the entire microbial genomes, as well as for comparing multiple reference genomes to mapped genomes of new organisms. Sequencing entire bacterial, viral, or other microbial genomes is important for the generation of accurate reference genomes, microbial identification, and other comparative genomic studies.
Compared with conventional approaches like PCR, Whole Genome Sequencing does not require labor-intensive cloning and mapping steps. Hence, it is time- and cost-effective. Moreover, this high-throughput sequencing approach allows the sequencing of numerous samples at the same time through the courtesy of multiplexing.
Applications of Microbial Whole Genome Sequencing
- Permits the detection of variations within target genomes
- Interpretation of character differences
- Allows large-scale evolution research
- Enables prerequisite study of novel species identification
Benefits of Microbial Whole Genome Sequencing
- Extensive experience: We have successfully completed high-profile projects which cover a wide range of fields such as pathogenic bacteria, probiotics, edible bacteria, medicinal strains, and industrial strains.
- Professional services: From material selection, library construction, and sequencing to data analysis, each step provides scientific and meticulous design to ensure high-quality research results.
- Comprehensive analysis: Detection of SNP, InDel, SV and other mutation information of strain reference genomes, and further research on species evolution, population characteristics, selection pressure, etc. One-stop analysis of variation and difference.
- Strict quality control: High-quality of the sequencing data is ensured by verifying the samples.
- High-quality library preparation: To ensure the quality of the data, all libraries are size selected to optimize the size of the insert.
eBooks

Applications of Long-Read Sequencing in Microbial Genomics
Determining the genomic sequences of microorganisms has an extraordinary potential for commercial applications and the advancement of scientific knowledge. These organisms exist in nearly all environments including soil, water, air, inside and among symbionts, and within hosts. To fully exploit and commercialize microbes’ unique potential, researchers capitalize on recent advances in DNA sequencing technology to determine the complete genome sequence of microbes composing entire populations and thus are able to identify the genes that govern all of their important processes as well as determine the compositions of complex bacterial communities.
Microbial Whole Genome Sequencing Specifications: DNA Sample Requirements
| Library Type | Sample Type | Amount (Qubit) | Volume | Concentration | Purity (NanoDropTM/ Agarose Gel) |
| Microbial whole genome library | Genomic DNA | ≥200 ng | ≥20 μL | ≥10 ng/μL | A260/280=1.8-2.0; no degradation, no contamination |
| Microbial whole genome library (PCR-free) |
Genomic DNA | ≥1.2 μg | ≥20 μL | ≥10 ng/μL |
Microbial Whole Genome Sequencing Specifications: Sequencing Parameters and Analysis Contents
| Platform Type | Illumina NovaSeq 6000 |
| Read Length | Paired-end 150 bp |
| Recommended Sequencing Depth | ≥ 100x for bacterial genomes |
| ≥ 50x for fungal genomes | |
| Standard Data Analysis |
|
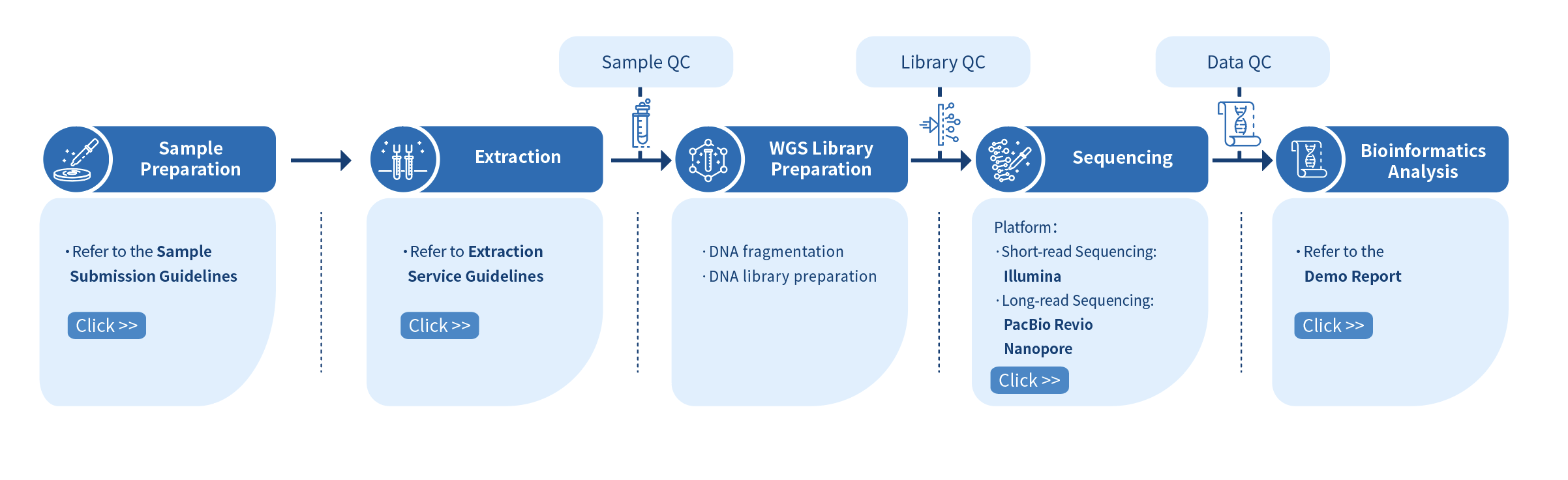
Novogene Workflow of Microbial Whole Genome Sequencing Service
The first step of the project workflow involves the sample quality control (Sample QC) to ensure that your samples meet the criteria of the Microbial WGS technique. Then, the appropriate library is prepared according to your target organism and subsequently tested for its quality (Library QC). Next, a paired-end 150 bp sequencing strategy is used to sequence the samples and the resulting data go through quality data control (Data QC) to guarantee the quality of the resulting data. Finally, bioinformatics analyses are performed and publication-ready results are provided. The following flowsheet describes the step-by-step protocol our Microbial WGS technique follows.
Preparation of sample is followed by the DNA library preparation which is verified for quality and yield. Genomic DNA is fragmented and size selected. The selected fragments are then end polished, A-tailed, and ligated with the full-length adapter. Illumina PE150 technology is employed to sequence the sample and the final stage involves the bioinformatics analysis.
Featured Publications of Microbial Whole Genome Sequencing
-
Dynamics and Microevolution of Vibrio parahaemolyticus Populations in Shellfish Farms
mSystems Date: 12 January 2021IF: 6.663DOI: https://doi.org/10.1128/mSystems.01161-20
-
Science of the Total Environment Date: 20 june 2021IF:6.551DOI: https://10.1016/j.scitotenv.2021.145767
-
food chemistry Date: 15 November 2020IF: 6.306DOI: https://10.1016/j.foodchem.2020.127316
-
Whole genome sequence of Diaporthe capsici, a new pathogen of walnut blight
Genomics Date: 23 February 2021IF: 6.205DOI: https://doi.org/10.1016/j.ygeno.2020.04.018
-
Effect of steel slag in recycling waste activated sludge to produce anaerobic granular sludge
Chemosphere Date: 25 October 2020IF: 5.108DOI: https://doi.org/10.1016/j.chemosphere.2020.127291
-
Journal of Global Antimicrobial Resistance Date: 23 December 2020IF: 4.035DOI: https://10.1016/j.jgar.2020.08.002
-
Alterations of gut microbiota contribute to the progression of unruptured intracranial aneurysms
Nature Communications Date: 25 june 2020IF:14.919DOI: https://10.1038/s41467-020-16990-3
SNP Mutation Frequency
Single nucleotide polymorphism (SNP) refers to a variation in a single nucleotide that can occur at some specific position in the genome, including transition and transversion of a single nucleotide.
Taken the T: A>C: G mutations as an example, this category includes mutations from T to C and A to G. When T>C mutation appears on either of the double-strand, the A>G mutation will be found in the same position of the other chain. Therefore the T>C and A>G mutations are classified into one category. Accordingly, the whole genome SNP mutations could be classified into six categories. The frequency of each type is shown in Figure X.
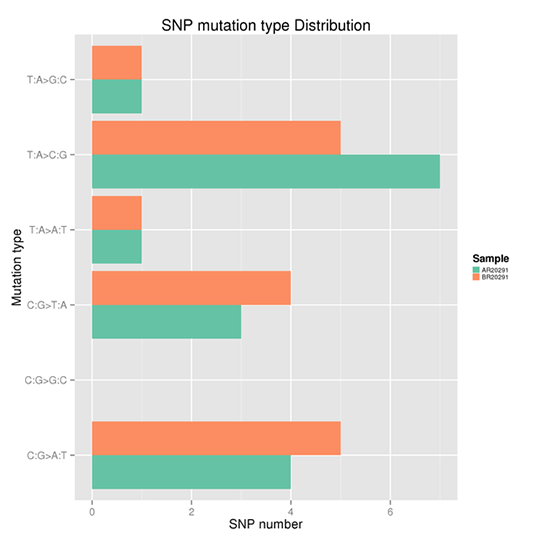
Note:
The x-axis represents the number of SNPs and the y-axis indicates the mutation types.
Length Distribution of CDS-located InDels
InDel refers to the insertion or deletion of ≤ 50 bp sequences in the DNA. The results demonstrate several peaks present at certain InDel lengths. Non-frameshift InDels exert a smaller effect on the genome as compared to frameshift InDels.
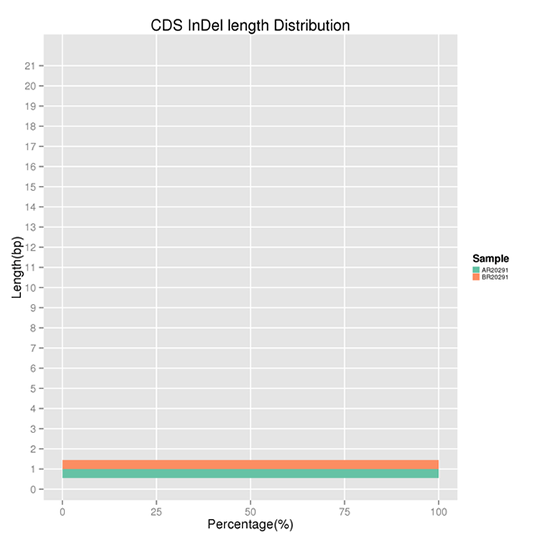
Note:
The x-axis represents the proportion of the InDels with a certain length, and the y-axis indicates the length of the InDels.
Length Distribution of SVs
Structural variants (SVs) are genomic variations with mutations of relatively larger size (>50 bp), including deletions, duplications, insertions, inversions, and balanced translocations.
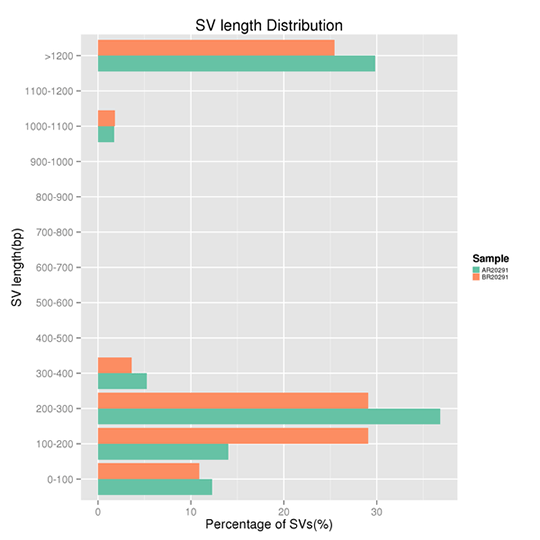
Note:
The x-axis represents the proportion of the SVs with a certain length range, and the y-axis indicates the certain length range of the SVs. Note, the length of DNA insert in library construction impacts the SVs detection greatly.
The distribution of CNVs on the genome
Copy-number variation (CNV) is a type of structural variation that happens when a DNA fragment is present in variable copy number in comparison to a reference genome. It pinpoints the deletions and duplications in the genome.
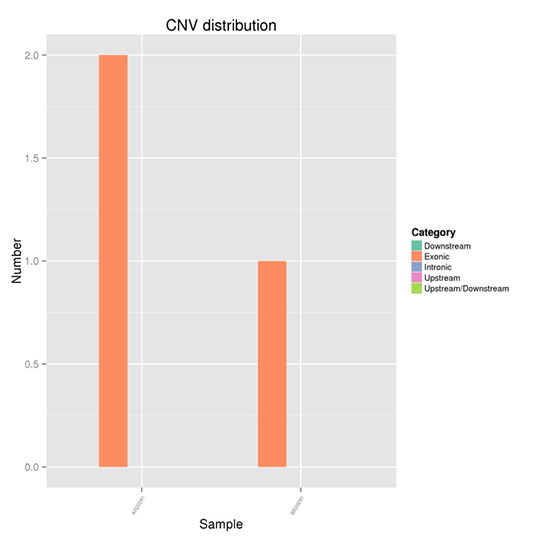
Note:
The x-axis represents samples and the y-axis indicates the number of CNVs in a different region.
Visualization of Variation
For proper visualization of the structural variations in the whole genome, we present mutation types with Circos:
(1) for SNP/InDel type, the density distribution is drawn;
(2) for SV/CNV type, the location and size are drawn.
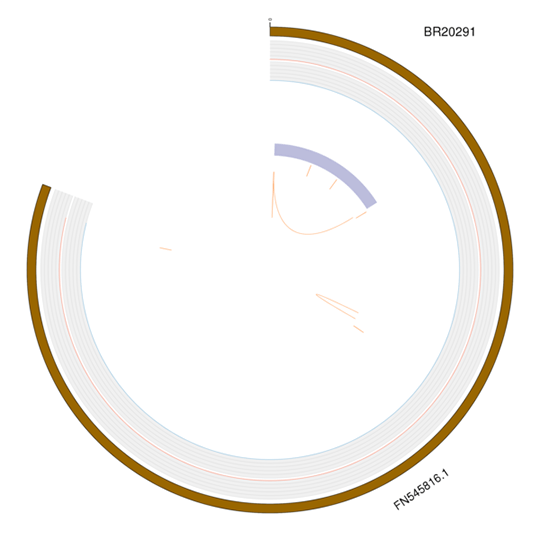
Note :
From outer to inner: chromosome, SNP, InDel, CNV duplication, CNV deletion, SV insertion, SV deletion, SV invertion, SV ITX, SV CTX.
*Please contact us to get the full demo report.